Genomic sequencing intro
Abbreviations
BWA (Burrows-Wheeler transformation aligner), FDR (False discovery rate), GO (Gene Ontology), GrCh38 (Genome Reference Consortium Human Build 38), GVCF (Genomic Variant Call Format), KEGG (Kyoto Encyclopedia of Genes and Genomes), LoF (loss-of-function), NCBI (National Center for Biotechnology), Pfam (Protein families database), PPI (Protein-protein interaction), VCF (variant call format).
Introduction
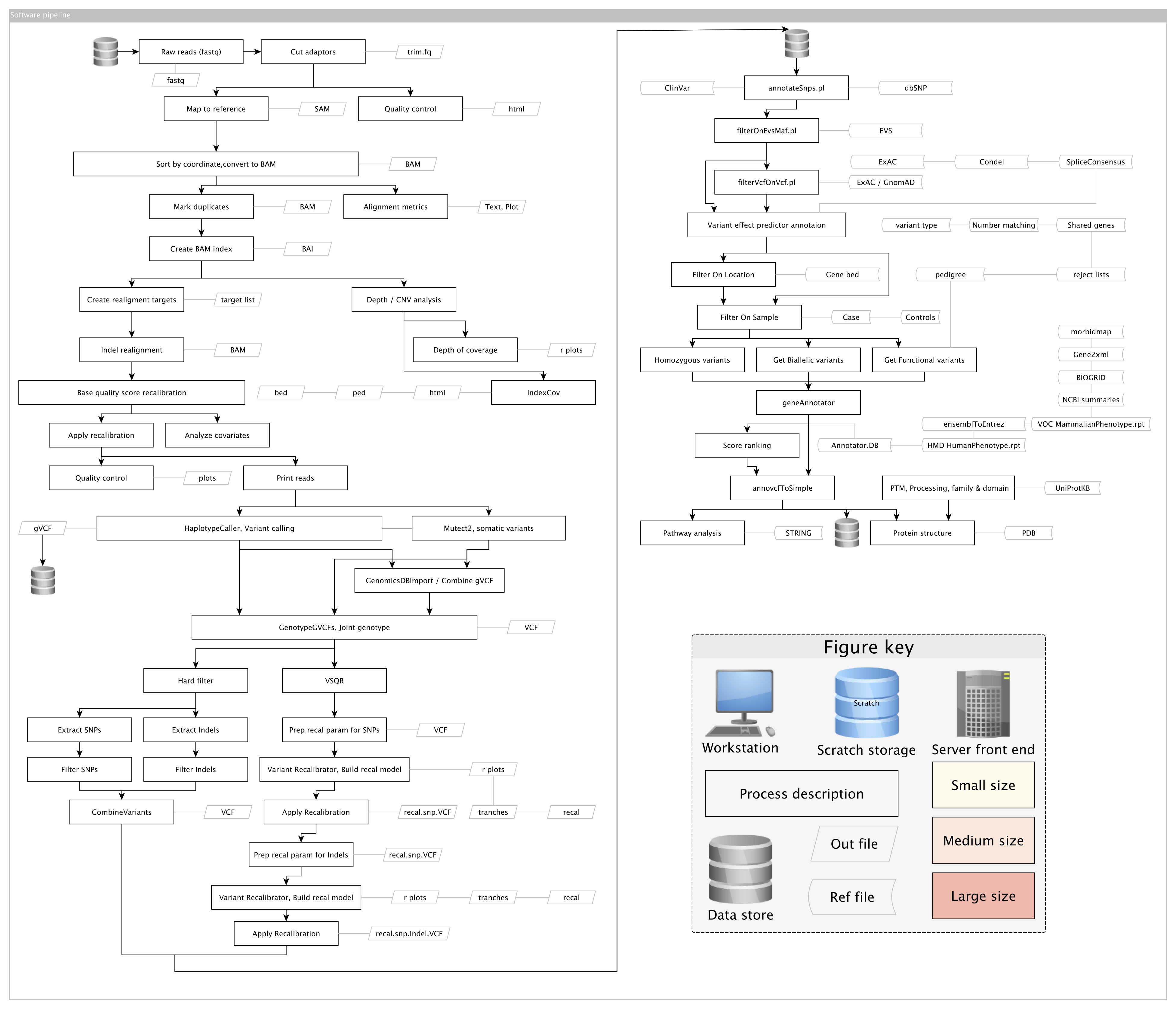
This overview contains theory and examples for the investigation of rare disease by exome sequencing. Each section is generally self-contained with a brief introduction. An example of a summarised analysis plan is illustrated is the following map. Heavy boxes represent bioinformatic protocols and slanted boxes represent key file types. Other reference file types are shown by arrow boxes.
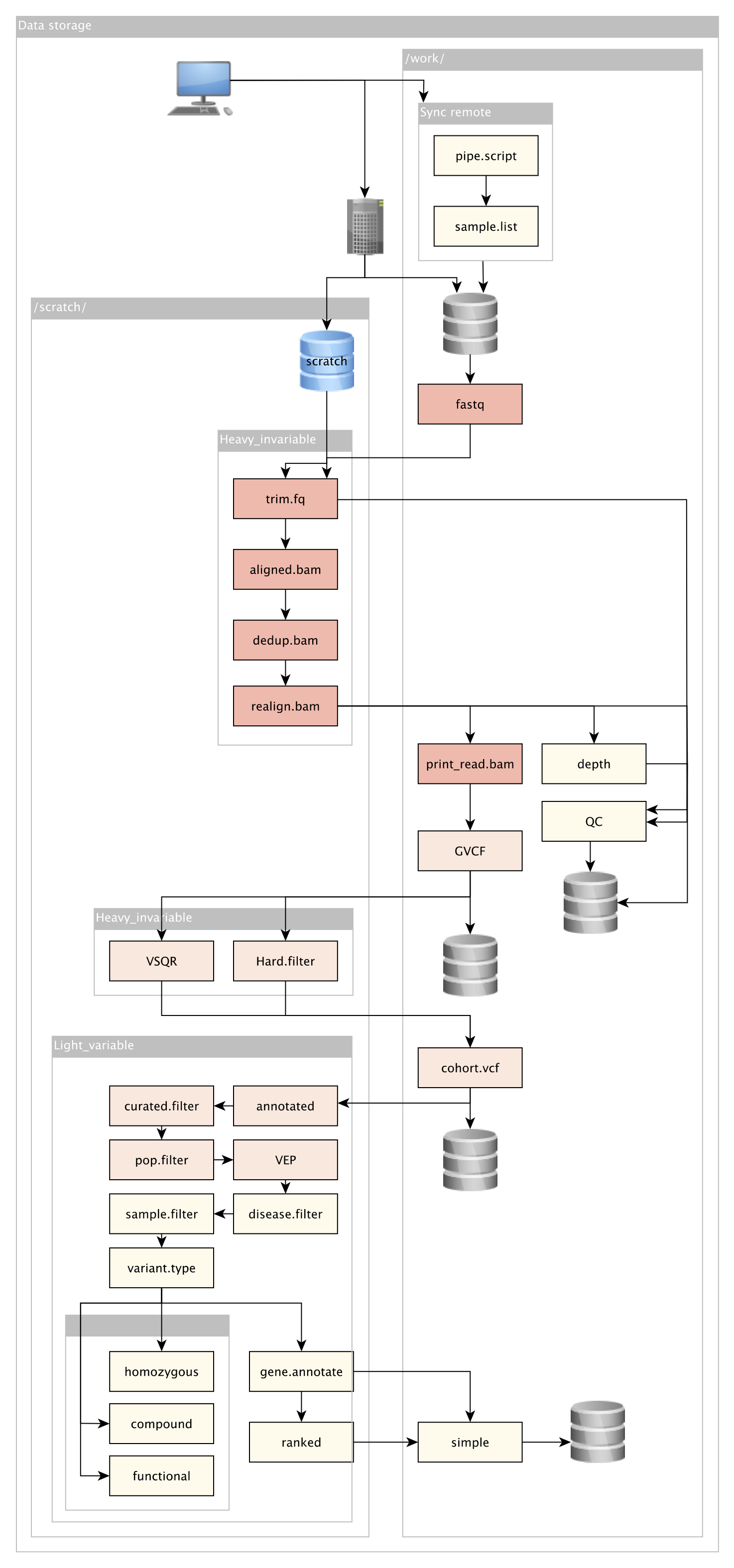
An example accompanying data storage plan can be seen here. A rough overview “infographic” of a next generation sequencing study is shown below. The general requirements, personnel responsibilities, and cost-breakdown is shown.
{kind=link}
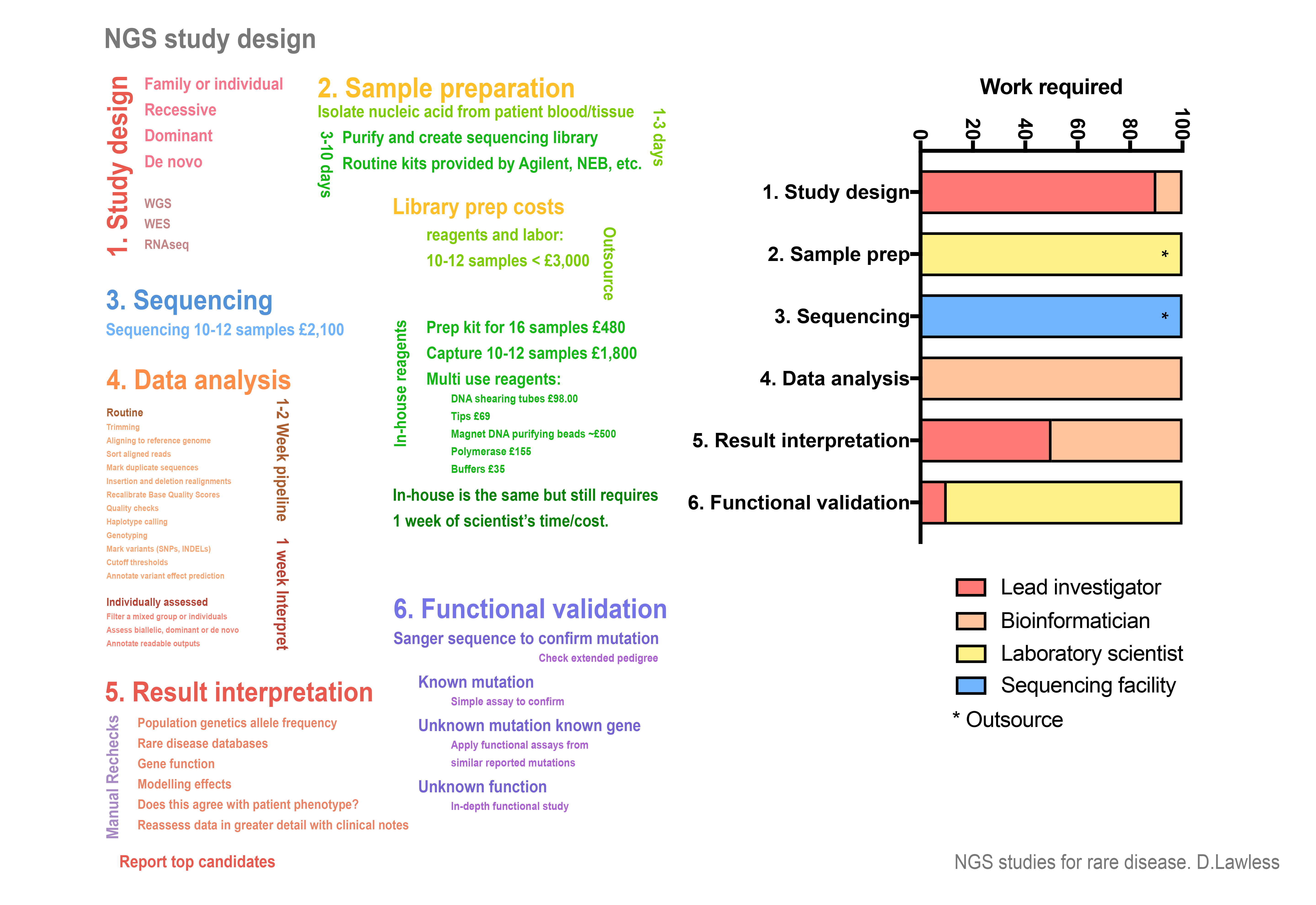
Whole exome sequencing experiment design. The general requirements, personnel responsibilities, and cost-breakdown is shown for a small NGS study of approximately ten participants. If library preparation and sequencing is performed at a dedicated facility then scaling up to very large cohorts (>1,000) potential only differs in one critical feature; implementing the bioinformatic methods used in this chapter also requires a critical expertise in high-performance computing. No methods have been included to demonstrate job scheduling and parallelisation across large computer clusters.
Exome sequencing
Sample preparation
For genomic investigations, a patient generally donates a small blood sample (2-6mL) along with signed consent to use their biological material and data in genetic and functional research. A small sample of hair or saliva are also succificient as a source of DNA if a blood sample is unavailable. Ideally, patient DNA is purified from peripheral blood monocytes. In most cases, the purification is done using a commercial kit such as that from Qiagen (51104 QIAamp DNA Blood Mini Kit). This protocol takes about 1 hour to purify 1-10 patient samples. Sometimes patient DNA is provided from an external source such as a local hospital where blood samples are processed routinely by dedicated staff. In this case, the purification method may be unknown so extra care should be taken when checking suitability for sequencing experiments. Consideration should be given to the possibility of sample mix up, that contamination could have occurred, etc.
High-throughput sequencing experiments benefit from consistency during sequencing library preparation. While there are several commercial options available, the protocol used in this study was the SureSelect XT target enrichment system for Illumina paired-end multiplexed sequencing library. A detailed protocol is available from the manufacturer. However, the process can be summarised in four main steps. After DNA quality has been checked, the basic protocol consists of:
- DNA fragmentation into 100-300 base pair strands, either
- (i) by using an enzyme that digests the DNA or
- (ii) by breaking by sonication; the DNA is suspended inside a small glass tube containing a glass rod which is vibrated by sonic waves inside a water bath.
- Another round of quality control checks to ensure that the DNA is fragmented into the correct size range.
- These fragments are bound by probes that specifically recognise the coding sequences which collectively make up the exome.
- The DNA that has been selectively purified is then tagged by adding a tail of nucleotides in specific sequences that label each of the individual samples with a unique code. When the sequencing step is performed later, all of the samples will get mixed together. The unique tag allows us to later re-identify which sequences belong to every person included in the study.
While it is important that library preparation is performed accurately, the individual steps could be replaced by alternative methods. The crucial component is an end product of targeted DNA fragments that have been tagged appropriately to allow the sequencing chemistry on the chosen system and that fragment lengths are in the correct range. A more detailed summary of the procedure is outlined;
Preparation of sample
- DNA is sheared, the most frequently used methods are by enzymatic digestion and sonication.
- Fragmented DNA is purified using AMPure XP beads.
- Quality assessment.
- End repair.
- Purify using AMPure XP beads.
- Adenylation at 3’ end.
- Purify using AMPure XP beads.
- Paired-end adaptor ligation.
- Purify using AMPure XP beads.
- Amplification.
- Purify using AMPure XP beads.
- Assess quality.
Hybridization and capture
- Hybridize capture library probes to DNA.
- Capture the hybridized DNA using streptavidin-coated beads. Note: at this step, custom gene target libraries can be used.
Indexing and multiplexing
- Captured libraries are amplified with indexing primers.
- Purify using AMPure XP beads.
- Assess quality and concentration of indexed library DNA.
- Pool samples at equal concentrations.
Capture library
For targeted sequencing experiments, the most important step in library preparation is the hybridization of capture library probes. Libraries of probes that are complementary to exome coding sequences can be ordered from a number of commercial suppliers. For a whole exome, this consists of hundreds of thousands of short RNA oligonucleotide strands bound to biotin. When the capture library hybridization mix is added to the DNA, most of the short probes bind to their complementary DNA sequences over 12-16 hours. To separate these selected fragments from the remaining bulk of unwanted DNA, streptavidin-coated magnetic beads are added. The streptavidin attaches to the biotin and therefore the DNA-bound probe can be pulled out using a strong magnet. Unbound DNA can then be washed away. Experiments in this study have been performed using Agilent capture library SureSelect Human All Exon V4-6, although several other options are available.
Targeted panels can also be used to focus on smaller sets of genes. For example, in some immunological conditions a panel of 50 genes might be targeted rather than a library for all known genes (exome). Cancer genetics screening services sometimes use a small panel of 40-100 genes. These small panels cut down on cost and focus only on genes where interpretation of variants would be possible. For the same price as whole exome, less capture library is needed and more samples can be sequenced.
As of 2018, all-exon capture library costs roughly £16,000 for enough reagent to prepare 96 DNA samples. This accounts for about 50% of the cost of the total library preparation materials. In total, the library preparation costs about £200 per sample. Once the samples have been prepared it cost about another £200 to sequence; approximately £400 total.
Sequencing
The sequencing carried out in this study was performed on Illumina platforms. These include the MiSeq for very small runs of a select set of genes, HiSeq 3000, 4000, and HiSeq X for whole exome or whole genome sequencing. The prepared libraries of patient DNA are pooled to contain 5-12 samples per pool. Since each sample has a unique identifier tag, it is OK to pool them together and later separate out all the individual data per person. On the HiSeq 3000 approximately 12 samples can be run per lane with acceptable coverage. This provides about 30-50X reads per nucleotide, sufficiently deep to confidently identify true germline mutations. There are 8 lanes per sequencing flow cell. Therefore, a single sequencing run can contain anything from 50-100 patient samples. Depending on the sequencing platform the run can take up to 5 days to complete.
Ultra-deep sequencing
Mendelian disorders can be successfully explained using exome and whole genome sequencing. Both the interpretability and cost per sample are improved in cases where a gene sequencing panel can be used. Some conditions, particularly autoinflammatory disorders, can arise from low frequency somatic variants that are capable of driving disease through potent gain-of-function mechanisms. It is worth noting that a “gain-of-function” can also be considered as a succinct description for systems where a loss of inhibitory activity occurs that directly results in increased signaling cascade activity that would otherwise rest in an inactive state; a homeostatic pathway. E.g. loss of an autoinhibitory feature for a single protein or loss of an inhibitory mechanism that is responsible for direct repression in the absence of stimulation or specific agonist. In such cases, a low frequency de novo variant will escape detection with typical sequencing methods, but ultra-deep sequencing offers a method for detection. This option uses a high concentration of capture reagent to prepare a highly enriched library and sequence at high-density on a flow cell to produce ultra-deep sequencing reads (e.g. >5,000x versus 50x, as typical for whole exome sequencing). In this case, PCR-free preparation is ideal for somatic variant detection, naturally.
Maintained by Dylan Lawless. Scientist at EPFL. Resume and Google scholar. RSS feed