Tailored analysis
Routine analysis typically takes up to a week, although it is usually performed on a standardised pipeline that can run automatically on a high-performance computing platform. A large part of custom filtering begins when the routine analysis steps have been completed; downstream analysis is adapted for each particular challenge. The discussion in explains some foundational steps towards a fully automatic system that relies only on some input features, such as clinical information. While many software packages exist that claim to output tailored analysis, these tend to either only tackle a specific niche or require lots of curated auxiliary input data.
The output of non-routine analysis (outlined in this chapter) sometimes takes only five minutes to interpret a cause of disease. In other cases, data that has been sequenced years previously has not yet relented an explanation for phenotypes that almost certainly should be explained by coding variants present within the sequence data. For example, for a dozen patients who share a similar and severe phenotype, it is often likely that the same gene or related genes could cause their disease. Unrelated patients with a rare dominant disease are not to all carry the same disease-causing variant; they may have different variants in shared gene, or variants among different genes which all contribute to a shared pathway that would result in the same end-point phenotype.
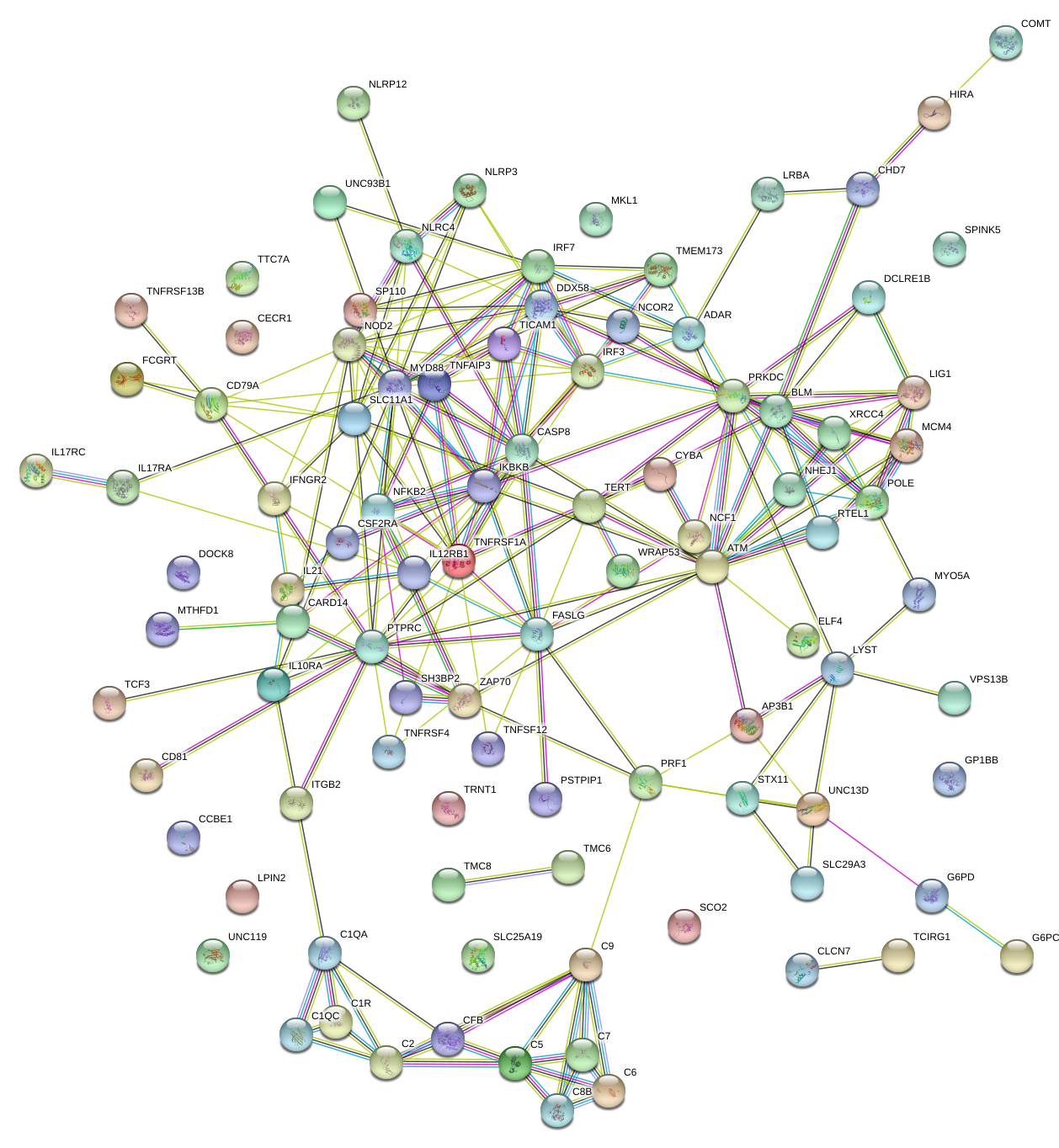
Shared pathways in candidate genes. Output of a STRING database query for known and predicted protein-protein interactions. These interactions include physical and functional associations. (STRING is an SIB-run service of the ELIXIR core data open resources for publicly funded research).
FigureLabel
string_normal_image
image_caption
For example, in Figure [fig:string_normal_image] above we see that from a group of unrelated people, all of the candidate genes carrying functional variants are joined by their shared functional interactions. For an autoinflammatory phenotype, genes like NLRP3, NOD2, TNFAIP3, MyD88, IKBKB, FASLG, or TMEM173 might all have different functions but damaging mutations in any of these could result in phenotypes that, on the surface, appear related.
Another circumstance might be seen in a small cohort of patients with a shared autoinflammatory phenotype. For example, the gene NLRP7 has relatively few publications examining it’s role in autoinflammatory disease. One would not consider this a strong candidate gene if faced with a variant of unknown significance in this gene from a single patient. However, three or four very rare or novel mutations in unrelated patients should be given consideration as producing an autoinflammatory disease. Single case, or small cohorts lack the power to measure significant associations. Therefore in the situation proposed here, manual interpretation is required (biased as it may be).
NLRP7 variants not reported as producing disease, like MEFV, or TNFAIP3. However, we must consider that genes plausibly responsible for causing disease in a dominant manner and that are highly conserved are generally under purifying selective pressure. Damaging mutations may be not be compatible with viability and therefore we never see cases of disease. Variants which are damaging to protein function but that do not completely destroy all of the normal structure may produce a phenotype that is pathogenic but viable with modern medical intervention.
In the example of NLRP7, the protein is known to Inhibit CASP1/caspase-1-dependent IL-1$\beta$ secretion. The functional domains of this protein are shared in other pro-inflammatory processes. Pyrin, NACHT, and LRR, domain variants are all studied for autoinflammatory diseases. The related gene, NLRP3, is probably the most widely recognised gene where damaging variants in these functional domains produce severe immune disorders. In cases where we have protein structures, we can also model the effect.
In our example, NLRP7 variants have been reported as the genetic determinant of a condition that causes early neonatal death and ectopic pregnancy. Many of the reported variants are stop mutations that will either produce a truncated protein or prevent expression of the allele altogether through nonsense-mediated decay. It is difficult predict the mechanism of disease in cases like this where the two outcomes have opposing paths. That is to say, a truncated protein may have an active functional domain which can no longer be inhibited since the C-terminal domains are missing, while haploinsufficiency would mean that cells cannot perform their normal function for the pathway since 50% of the protein is depleted (in heterozygous cases). Haploinsufficiency can result in a disease that phenotypically resembles a gain-of-function when the responsible protein normally acts as an inhibitor for an inflammatory pathway (Lawless et al., 2018). This is not expected with NLRP7 and therefore heterozygous loss-of-function does not explain disease.
For a candidate gene like this, we have some plausible evidence but cannot really progress any further without new functional studies. The first step involved confirmatory Sanger sequencing for all patients identified through exome sequencing. Next, any close relatives that are available might be also sequenced for the same variants. If the mutations are disease-causing then other carriers would also be expected to have some shared phenotype features. The possibilities in functional experiments vary widely and are highly dependent on the candidate genes. The procedure outlined in this hypothetical example is generally applicable in for the majority of single-case studies and illustrates the importance of tailored analysis. The initial findings of genomic analysis may produce more follow up questions, including whether other probable gene candidates can be ruled out, for which the patient carries only the “normal” reference alleles (e.g. CFTR screening for cystic fibrosis/lung disease).
References
- Lawless, D., Pathak, S., Scambler, T. E., Ouboussad, L., Anwar, R., & Savic, S. (2018). A Case of Adult-Onset Still’s Disease Caused by a Novel Splicing Mutation in TNFAIP3 Successfully Treated With Tocilizumab. Frontiers in Immunology, 9. https://doi.org/10.3389/fimmu.2018.01527
Maintained by Dylan Lawless. Scientist at EPFL. Resume and Google scholar. RSS feed