Quantification of pLoF
Identification of pLoF variants
As reported in “Sequencing of 53,831 diverse genomes from the NHLBI TOPMed Program” [0], predicted loss-of-function (pLoF) variants can be identified using Variant Effect Predictor (VEP) [1] and Loss Of Function Transcript Effect Estimator (LOFTEE) [2], with accurate genomic coordinates from GENCODE [3]. An enrichment of share biological function in affected genes can then be tested using Gene Ontology (GO) [4].
A note here about the pLoF acronym. I believe the “p” was originally for predicted loss-of-function, and has more recently changed to putative loss-of-function. The former is more transparent and preferable, I believe.
Variant Effect Predictor
Generally a local version of
Variant Effect Predictor
(VEP) software and databases are run for analysis.
However, one can try out the online version using the ensembl VEP URL.
An example output is shown here:
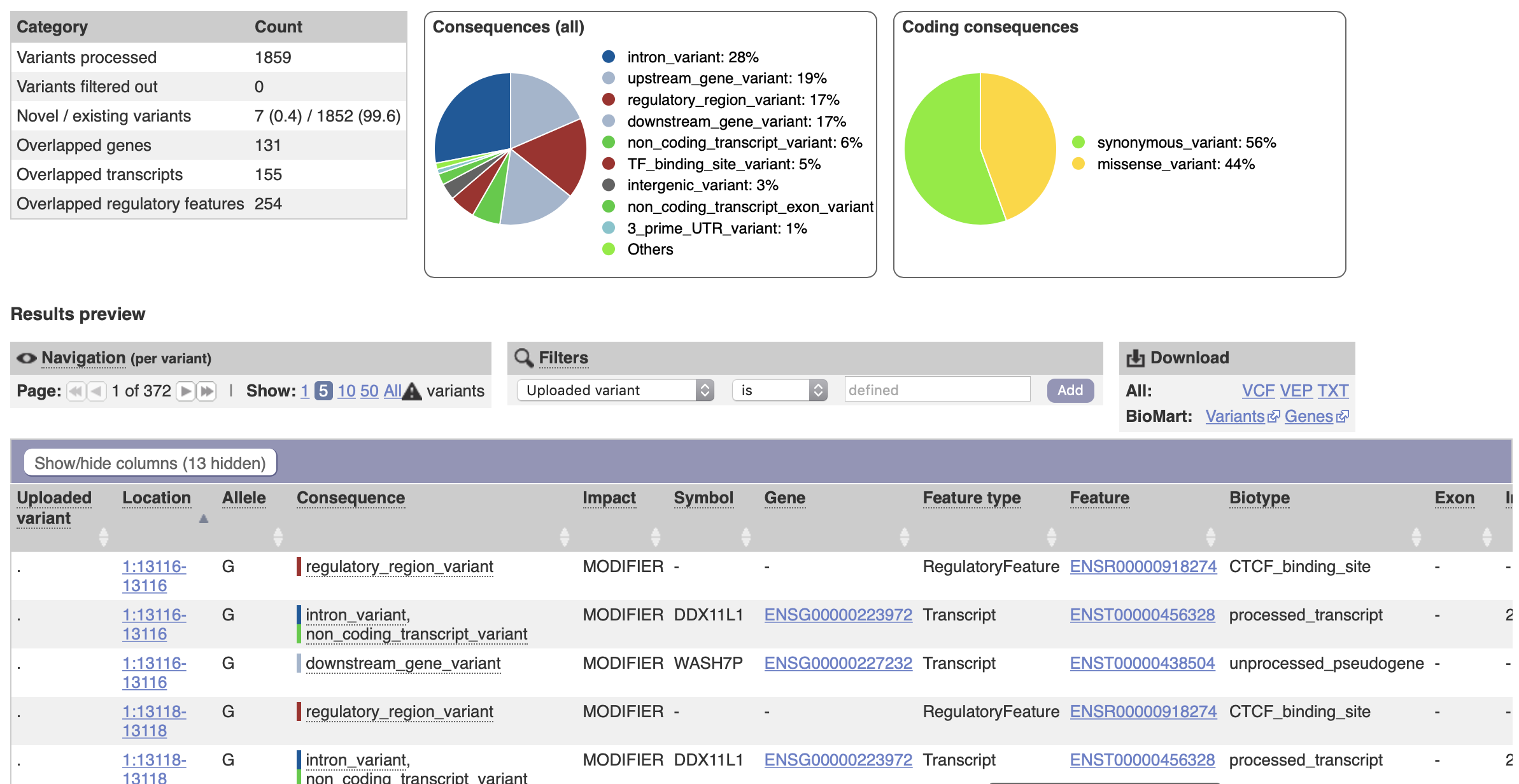
An obvious error that can occur is using mismatched genome builds.
Make certain that you use the same reference genome as used on the input data.
The Ensembl/VEP website URL should be for the same genome build (GRCh37, or the default GRCh38).
Local software and databases will also use the appropriate versions.
In variant annotation, VEP supplies a “consequence” column.
Consequences are general and based on translation of genetic code in humans.
The Loss-of-function (LoF) consequence is the simplest example (splice, stop mutations).
For the topic of variant collapsing, used in areas such as burden testing,
variant consequence defines which variants can be included in analysis since they are interpretable or of ostensibly known significance [4].
This could introduce spurious results so it is best to have a solid criteria for selecting consequences of interest.
The consequences provided by VEP are too long to discuss in detail.
The table from the ensembl website is worth reading:
Ensembl Variation - Calculated variant consequences.
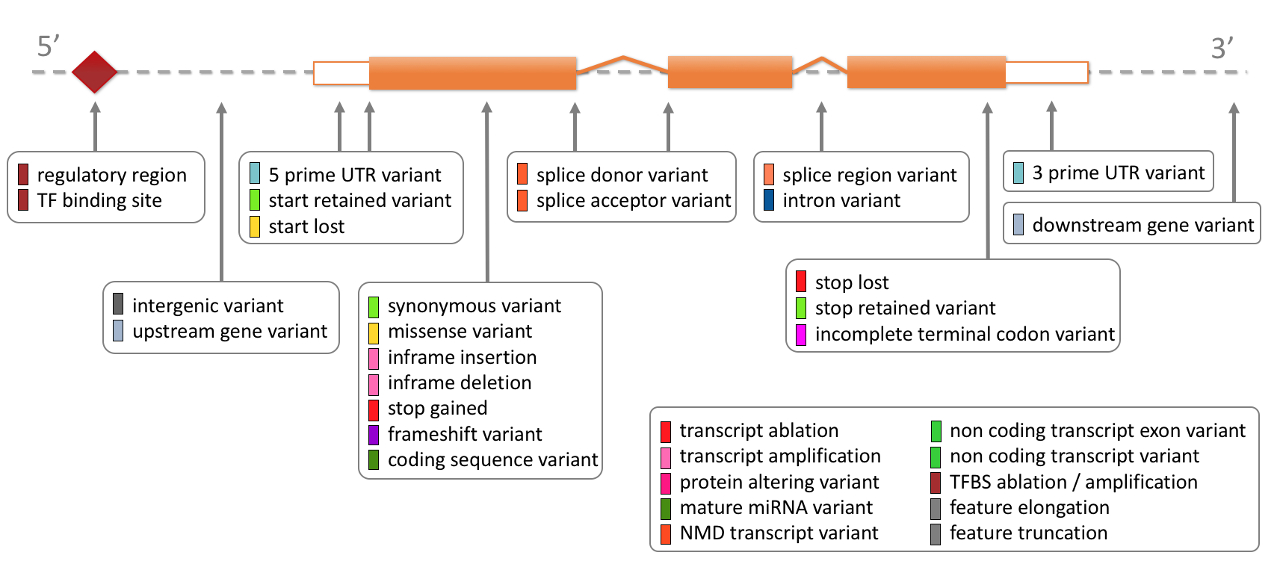
LOFTEE
The LOFTEE github repo shows the code and examples of usage. It functions as a VEP plugin for 3 LoF consequences:
- stop-gained
- frameshift variants
- splice site disrupting
There are some additional smart features that make LOFTEE more useful than simply the VEP consequences. These include known features about how transcripts will be affected by variants (e.g. stop variants near the end of a transcript), known splicing sequence mechanisms, and other flags including splicing prediction that may be less reliable but still valuable.
GENCODE
Accurate annotation depends on correctly mapping positions based on the genome reference. The genomic coordinates of coding elements may be based on GENCODE [3].
Gene Ontology (GO)
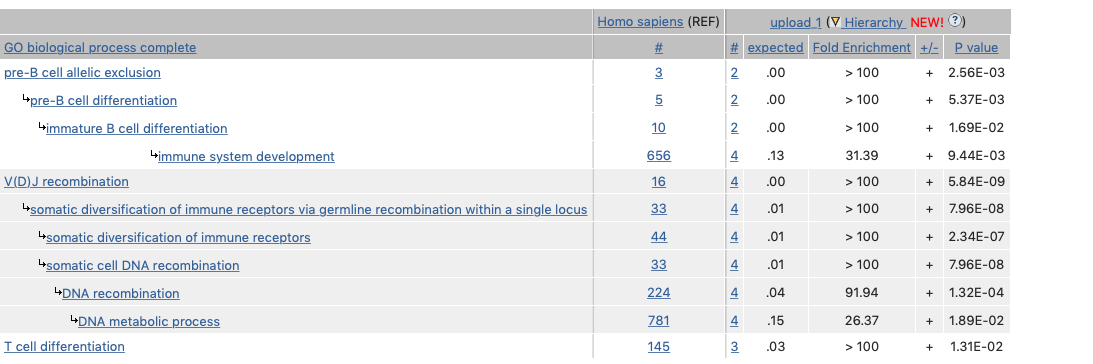
Gene Ontology (GO) is a valuable database of known gene function and can also be used to perform enrichment analysis on gene sets [5, 6]. For example, given a set of genes that are up-regulated under certain conditions, an enrichment analysis will find which GO terms are over-represented (or under-represented) using annotations for that gene set. This description was taken directly from geneontology.org where a longer explanation is shown. Using the web interface, an example query for 4 genes from the VDJ recombination pathway were queried. The output is shown here. The reference database has 20595 IDs. My 4 gene IDs were then tested for all known interactions - the number of shared GO terms. There were 9038 terms checked in total. The output table shows the GO process “V(D)J recombination” as the strongest association;
- all 4 genes shared this process
- for 4 random genes we would expect none to share the same GO process
The fold enrichment and strong P-value indicate a true association of shared biological pathway.
Published example
Methods
In Taliun et al [0], each of these tools are used in their analysis to target variants:
- stop-gained
- frameshift
- splice-site-disturbing variants
- annotated as high-confidence pLOF
- remove pLOF with allele frequency > 0.5%
- remove pLOF within regions masked due to poor accessibility
These last two additionally ignore variants that are likely of minor consequence to human health but still passed the basic pLoF filter. Taliun et al. evaluated enrichment and depletion of pLOF variants (allele frequency $<$ 0.5%) in gene sets (that is, terms) from Gene Ontology (GO) [5, 6]. For each gene annotated with a particular GO term, they computed the number of pLOF variants per
- protein-coding base pair, L, and
- proportion of singletons, S.
They then tested for lower or higher mean L and S in a GO term using bootstrapping (1,000,000 samples) with adjustment for the gene length of the protein-coding sequence (CDS):
- Sort all genes by CDS length in ascending order and divide them into equal-size bins (1,000 genes each);
- Count how many genes from a GO term are in each bin;
- From each bin, sample with replacement the same number of genes and compute the average L and S;
- Count how many times sampled L and S were lower or higher than observed values.
The P-values were computed as the proportion of bootstrap samples that exceeded the observed values. The fold change of average L and S was computed as a ratio of observed values to the average of sampled values. They tested all 12,563 GO terms that included more than one gene. The P-value significance threshold was thus ~2e-6. The enrichment and depletion of pLOF variants in public gene databases was tested in a similar way.
Results
Taliun et al. wanted to see if any gene sets had enrichment or depletion of rare pLoF. In the first 53,831 TOPMed samples, they detected more than 400 million variants:
- single-nucleotide variants
- insertions or deletions
Many novel variants will have been detected because they include
- assembly of unmapped reads and
- customized analysis in highly variable loci.
Among the more than 400 million detected variants,
- 97% have frequencies of less than 1% and
- 46% are singletons that are present in only one individual.
The 46% is pretty surprising but the number of private variant per person is probably not extremely high. In clinical exomes we usually expect approximately $<$100 novel variants compared to the in-house sequence database. We also expect aprrox. $<$10 de novo variants in the same sample if parents/family were also sequenced. The number will be much larger for non-coding genome, but since SNVs will be largely non-interpretable it is currently ignored for clinical diagnosis.
A notable class of variants is the 228,966 putative loss-of-function (pLOF) variants that were observed in 18,493 (95.0%) GENCODE genes (Extended Data Table 5 and Supplementary Fig. 12). This class includes the highest proportion of singletons among all of the variant classes that was examined. An average individual carried 2.5 unique pLOF variants. They identified more pLOF variants per individual than in previous surveys based on exome sequencing; an increase that was mainly driven by
- identification of additional frameshift variants (Table S6)
- and more uniform and complete coverage of protein-coding regions (Fig. S13, S14).
It is worth noting that the referenced figures (S13,14) don’t show specifically pLoF, but rather sequence depth per gene. They show that TopMed has better coverage/depth than ExAC, including for known disease genes. I would have expected them to show the same results specifically for pLoF variants to support their statement.
The extract from Table S7 shows the comparison of pLoF versus other rare variants in GO terms (protein pathways). The comparison is basically: Random sampling provides the number of expected variants, which is then compared to the observed number of variants. There are four categories used:
- expected pLoF
- observed pLoF
- expected singleton
- observed singleton
The top result, DNA-binding genes, showed rare variants (singletons) observed/expected = 1, but pLoF observed/expected = 0.5. This comparison is made to show that:
- This biological mechanism cannot tollorate damaging pLoF variants (pLoF/bp).
- And even though pLoF are usually very rare, this does not account for the lack of observations - rare/novel variants that are non-pLoF are present (proportion of singletons).
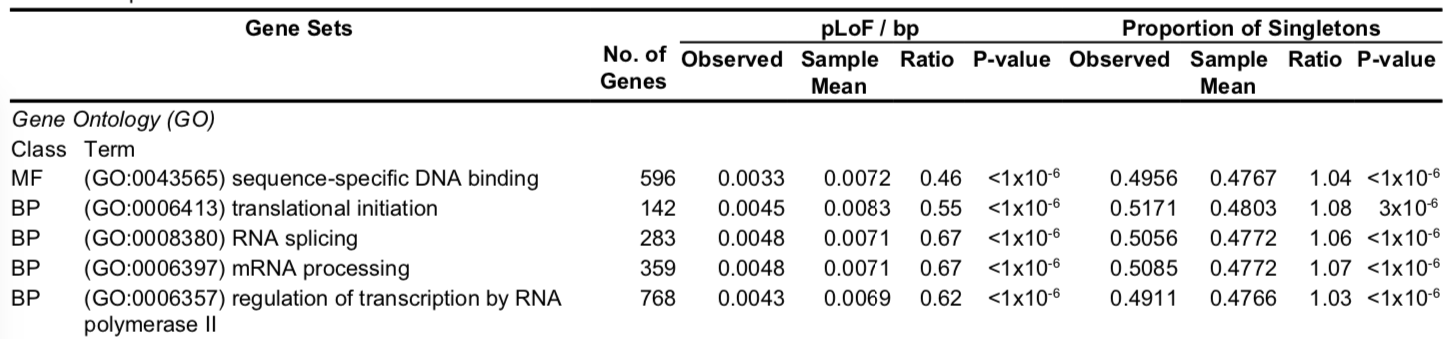
They searched for gene sets with fewer rare pLOF variants than expected based on gene size. The gene sets with strong functional constraint included genes that encode DNA- and RNA-binding proteins, spliceosomal complexes, translation initiation machinery and RNA splicing and processing proteins (Supplementary Table 7). They found fewer rare pLOF variants than expected (each comparison P $<$ 10−4) for Genes associated with human disease in
- COSMIC [7] (31% depletion),
- GWAS catalogue [8] (around 8% depletion),
- OMIM19 [9] (4% depletion) and
- ClinVar [10] (4% depletion).
The future
With very large scale genome sequencing we will start to association studies for extremely rare variants (eventually there will be no novel SNVs). This provokes a few possibilities.
- Predicting probably of all SNVs.
- CADD predicts pathogenicity for all SNVs but currently I don’t know of any mutation probability projects. Ratio of obs/exp will have valuable insight.
- All coding variants will be reported.
- functional prediction accuracies can be validated.
- Novel non-coding variants will become the new variants of unknown significance (VUS). Will we slowly trudge through the same system again or will there be a more sophisticated approach for the non-coding genome in clinical genomics?
References
[0] Taliun, Daniel, et al. Sequencing of 53,831 diverse genomes from the NHLBI TOPMed Program. Nature 590.7845 (2021): 290-299.
https://doi.org/10.1038/s41586-021-03205-y
[1] McLaren, W. et al. The Ensembl Variant Effect Predictor. Genome Biol. 17, 122 (2016).
[2] Karczewski, K. J. et al. loftee. GitHub https://github.com/konradjk/loftee (2015).
[3] Frankish, A. et al. GENCODE reference annotation for the human and mouse genomes. Nucleic Acids Res. 47 (D1), D766–D773 (2019).
[4] Povysil, Gundula, et al. “Rare-variant collapsing analyses for complex traits: guidelines and applications.” Nature Reviews Genetics 20.12 (2019): 747-759.
[5] The Gene Ontology Consortium. Gene ontology: tool for the unification of biology. Nat. Genet. 25, 25–29 (2000).
[6] The Gene Ontology Consortium. Expansion of the Gene Ontology knowledgebase and resources. Nucleic Acids Res. 45 (D1), D331–D338 (2017).
[7] Forbes, S. A. et al. COSMIC: exploring the world’s knowledge of somatic mutations in human cancer. Nucleic Acids Res. 43, D805–D811 (2015).
[8] Welter, D. et al. The NHGRI GWAS Catalog, a curated resource of SNP–trait associations. Nucleic Acids Res. 42, D1001–D1006 (2014).
[9] Hamosh, A., Scott, A. F., Amberger, J. S., Bocchini, C. A. & McKusick, V. A. Online Mendelian Inheritance in Man (OMIM), a knowledgebase of human genes and genetic disorders. Nucleic Acids Res. 33, D514–D517 (2005).
[10] Landrum, M. J. et al. ClinVar: improving access to variant interpretations and supporting evidence. Nucleic Acids Res. 46 (D1), D1062–D1067 (2018).\
Maintained by Dylan Lawless. Scientist at EPFL. Resume and Google scholar. RSS feed