Aggregated Cauchy Association Test (ACAT)
- Abbreviations
- Intro to this topic
- Papers
- Step-by-step explanation of ACAT.R
- Applying STAAR-O for multiple annotation weights
- Non-gene-centric analysis using dynamic windows with SCANG-STAAR
- Multi-weight annotation analysis
- Main equations for ACAT
- tan and \(\pi\)
- Original R code from yaowuliu
Abbreviations
- ACAT: Aggregated Cauchy Association Test
- ACAT-V: Aggregated Cauchy Association Test - Variant level
- ACAT-O: Aggregated Cauchy Association Test - Omnibus
- SKAT: Sequence Kernel Association Test
- ARIC: Atherosclerosis Risk in Communities
Intro to this topic
The Aggregated Cauchy Association Test (ACAT) is a statistical method used for rare-variant association tests (RVATs) in genetic studies. ACAT is designed to aggregate the association signals of multiple rare genetic variants within a genomic region or a gene, while accounting for the directions of the effects of these variants on the phenotype of interest. The ACAT method utilizes a Cauchy distribution, which allows for improved performance in identifying true associations, especially when the directions and magnitudes of variant effects are heterogeneous.
First, here is a great talk by author of SKAT and other methods:
Watch on YouTube Dr. Xihong Lin: Overview of Rare Variant Analysis of Whole Genome Sequencing Association Studies.
- The major part starts at time: 22:30
- and the best is at: 30:50 where she describes the aggregated Cauchy association test (ACAT) method for combining multiple annotations (like CADD score, MAF, etc.) to calculate the final P-value.
- This is their annotation database discussed: https://favor.genohub.org
Papers
- ACAT paper, Yaowu Liu, et al AJGH 2019.
- Application of STAAR protocol to TOPMed, Xihao Li, et al. NatGen 2020.
- STAAR pipeline methods paper, Zilin Li, et al. NatMethods 2022.
- STAAR pipeline github
- Controlling SKAT function: Here’s a summary of the SKAT package functions - which are easier to understand than reading the notation in the SKAT papers. If you read the code you see each new implement is added sequentially and how weights work. Although, the ACAT git repo is independent.
- ACAT git repo: https://github.com/yaowuliu/ACAT/blob/master/R/ACAT.R
Step-by-step explanation of ACAT.R
This discussion refers to code in the main ACAT function found at https://github.com/yaowuliu/ACAT.
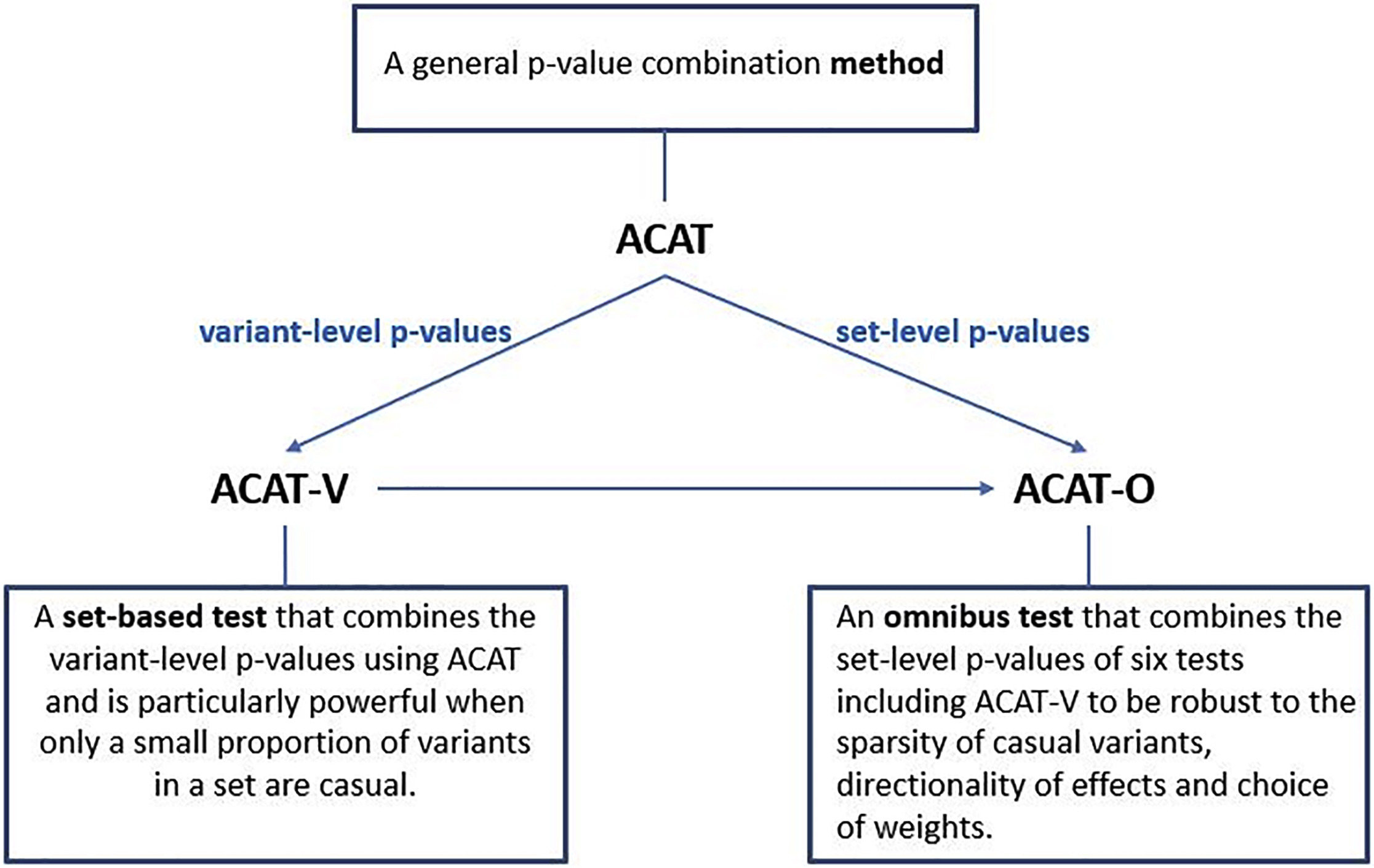
Figure 1. Summary of the Proposed Methods ACAT, ACAT-V, and ACAT-O and the Relationship Among Them. From the ACAT paper
- The R code defines several functions to perform the Aggregated Cauchy Association Test (ACAT) and the ACAT-V test.
-
ACAT: This function combines p-values using the Cauchy distribution. -
ACAT_V: A set-based test that uses ACAT to combine the variant-level p-values. -
NULL_Model: Computes model parameters and residuals for ACAT-V. -
Get.marginal.pval: A helper function to calculate the marginal p-values for ACAT-V.
-
-
ACATfunction- a. It accepts
Pvals(p-values),weights, and an optionalis.check parameterto validate the input. - b. Checks for NA, p-value range (0 to 1), and existence of both 0 and 1 p-values in the same column.
- c. If weights are not provided, equal weights are used. Otherwise, user-supplied weights are validated and standardized.
- d. The function calculates the Cauchy statistics and returns the ACAT p-value(s).
- a. It accepts
-
ACAT_Vfunction- a. It accepts
G(genotype matrix),obj(output object ofNULL_Model),weights.beta,weights, andmac.thresh. - b. It checks for the validity of input weights.
- c. Based on the
mac.threshvalue, it decides to use the Burden test, the Cauchy method, or a combination of both. - d. It calculates the final p-value and returns it.
- a. It accepts
-
NULL_Modelfunction- a. It accepts
Y(outcome phenotypes) andZ(covariates). - b. It determines if
Yis continuous or binary. - c. It fits a linear regression model if
Yis continuous and a logistic model ifYis binary. - d. It returns an object with model parameters and residuals.
- a. It accepts
-
Get.marginal.pvalfunction- a. It accepts
G(genotype matrix) andobj(output object ofNULL_Model). - b. It checks the validity of the
objinput. - c. It calculates the marginal p-values and returns them.
- a. It accepts
The Aggregated Cauchy Association Test (ACAT) is a powerful and computationally efficient method designed to improve the analysis of rare and low-frequency genetic variants in sequencing studies. Traditional set-based tests can experience power loss when only a small proportion of variants are causal, and their power can be sensitive to factors such as the number, effect sizes, and effect directions of causal variants, as well as weight choices.
ACAT addresses these issues by combining variant-level p-values to create a set-based test called ACAT-V. ACAT-V is particularly powerful when there are only a few causal variants in a set, making it a valuable tool for genetic analysis. Additionally, ACAT can be used to create an omnibus test called ACAT-O by combining different variant-set-level p-values. ACAT-O incorporates the strengths of multiple complementary set-based tests, such as the burden test, sequence kernel association test (SKAT), and ACAT-V.
By analyzing extensive simulated data and real-world data from the Atherosclerosis Risk in Communities (ARIC) study, it has been demonstrated that ACAT-V complements other tests like SKAT and the burden test. Furthermore, ACAT-O consistently delivers more robust and higher power than alternative tests, making it a valuable addition to the toolkit of researchers working with sequencing studies.
ACAT is designed to combine p-values from multiple variants or tests rather than combining annotation scores directly. If you have p-values associated with each of the 5 annotation columns (CADD_score, MAF, GnomAD_AF, REVEL_score, ClinVar_score) for a single variant, you could potentially use ACAT to combine these p-values to obtain a single combined p-value for that variant. However, it’s essential to ensure that the p-values are valid and independent for ACAT to be effective.
To do this see the STAAR framework for this.

Figure 2. Slide from presentation of ACAT method.
Applying STAAR-O for multiple annotation weights
In a separate page, I discuss the STAAR method. The following passages are included in both pages since they related.
In the STAAR Nature Methods paper, the section Gene-centric analysis of the noncoding genome shows how the STAAR method can indeed be used to capitalize on the ACAT method to obtain a combined p-value from a set of annotations for a single variant. The STAAR framework incorporates multiple functional annotation scores into the RVATs (rare-variant association tests) to increase the power of association analysis. In this context, it uses the STAAR-O test, an omnibus test that aggregates annotation-weighted burden test, SKAT, and ACAT-V within the STAAR framework.
By incorporating multiple functional annotation scores, such as CADD, LINSIGHT, FATHMM-XF, and annotation principal components (aPCs), the STAAR method enhances the ability to detect associations between variants and traits of interest. Therefore, the STAAR framework can be used to leverage the strengths of the ACAT method and obtain a combined p-value from a set of annotations for a single variant or a set of variants.
Non-gene-centric analysis using dynamic windows with SCANG-STAAR
The SCANG-STAAR method is an improvement over the fixed-size sliding window RVAT in the STAAR framework. It proposes a dynamic window-based approach called SCANG-STAAR, which extends the SCANG procedure by incorporating multidimensional functional annotations. This method allows for flexible detection of locations and sizes of signal windows across the genome, as the locations of regions associated with a disease or trait are often unknown in advance, and their sizes may vary across the genome. Using a prespecified fixed-size sliding window for RVAT can lead to power loss if the prespecified window sizes do not align with the true locations of the signals.
The SCANG-STAAR method has two main procedures: SCANG-STAAR-S and SCANG-STAAR-B. SCANG-STAAR-S extends the SCANG-SKAT (SCANG-S) procedure by calculating the STAAR-SKAT (STAAR-S) p-value in each overlapping window by incorporating multiple variant functional annotations, instead of using just the MAF-weight-based SKAT p-value. SCANG-STAAR-B is based on the STAAR-Burden p-value. SCANG-STAAR-S has two advantages over SCANG-STAAR-B in detecting noncoding associations using dynamic windows: first, the effects of causal variants in a neighborhood in the noncoding genome tend to be in different directions, especially in intergenic regions; second, due to the different correlation structures of the two test statistics for overlapping windows, the genome-wide significance threshold of SCANG-STAAR-B is lower than that of SCANG-STAAR-S.
SCANG-STAAR also provides the SCANG-STAAR-O procedure, based on an omnibus p-value of SCANG-STAAR-S and SCANG-STAAR-B calculated by the ACAT method. However, unlike STAAR-O, the ACAT-V test is not incorporated into the omnibus test because it is designed for sparse alternatives, and as a result, it tends to detect the region with the smallest size that contains the most significant variant in the dynamic window procedure.
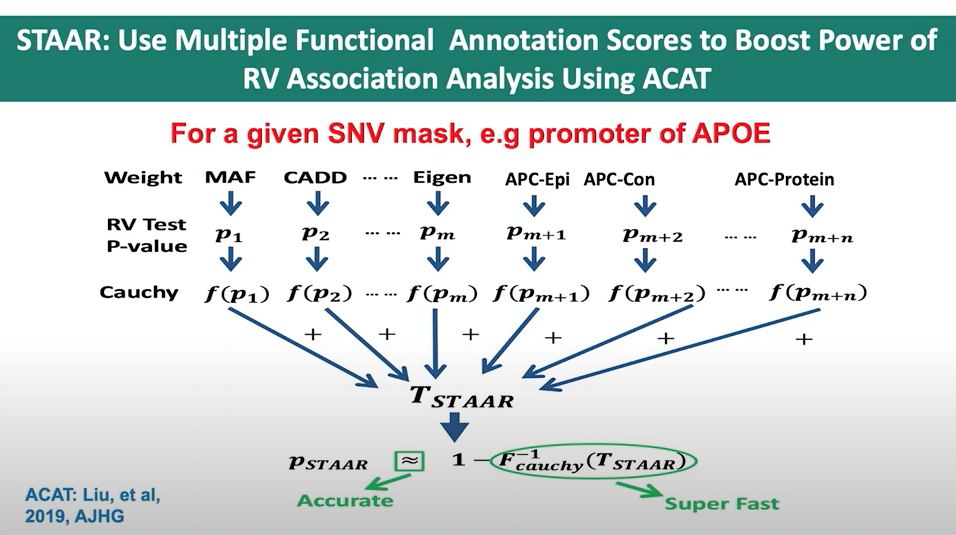
Figure 3. Slide from presentation of ACAT application in STAAR.
Multi-weight annotation analysis
The STAAR framework can be used to combine the p-values associated with each of the 5 annotation columns (CADD_score, MAF, GnomAD_AF, REVEL_score, ClinVar_score) for a single variant. STAAR incorporates multiple functional annotation scores as weights when constructing its statistics, making it suitable for combining p-values from different annotation columns to obtain a single combined p-value for that variant.
Main equations for ACAT
- ACAT test statistic:
where \(p_i\) are the p-values, and \(w_i\) are non-negative weights.
- P-value calculation for the ACAT test statistic:
where \(w = \sum_{i=1}^{k} w_i\).
- ACAT is a general and flexible method of combining p-values, which can represent the statistical significance of different kinds of genetic variations in sequencing studies.
- ACAT only aggregates p-values, so one can automatically control cryptic relatedness and/or population stratification by fitting appropriate models from which p-values are calculated through methods such as principal-component analysis or mixed models.
- The null distribution of the test statistic \(T_{ACAT}\) can be well approximated by a Cauchy distribution without the need for estimating and accounting for the correlation among p-values.
- Calculating the p-value of ACAT requires almost negligible computation and is extremely fast.
- The approximation is particularly accurate when ACAT has a very small p-value, which is useful in sequencing studies because only very small p-values can pass the stringent genome-wide significance threshold and are of particular interest.
tan and \(\pi\)
In the ACAT method, the “tan” and “π” functions are used to transform the p-values in such a way that they follow a standard Cauchy distribution under the null hypothesis. This transformation is essential to the ACAT method because it allows for an efficient and accurate combination of p-values, even when they are correlated.
The reason for using the tangent function (“tan”) specifically is because of its connection to the Cauchy distribution. The Cauchy distribution has some unique properties, such as having a heavy tail, which make it suitable for handling correlated p-values in this context. The transformation function used in the ACAT method, given by \(tan((0.5 - p_i) \pi)\), ensures that if the p-value \(p_i\) is from the null distribution, the transformed value will follow a standard Cauchy distribution.
The constant \(\pi\) (Pi) is used in the formula because it is a natural component of the tangent function. In the context of the ACAT method, \(\pi\) is used to scale the input of the tangent function, which is necessary to map the range of p-values (0 to 1) to the entire domain of the tangent function. This ensures that the transformed values will follow the desired Cauchy distribution.
Therefore, the “tan” and \(\pi\) functions in the ACAT method are used to transform p-values so that they follow a standard Cauchy distribution under the null hypothesis, which allows for an efficient and accurate combination of correlated p-values.
Original R code from yaowuliu
This code is the main ACAT function found at https://github.com/yaowuliu/ACAT
#'
#' Aggregated Cauchy Assocaition Test
#'
#' A p-value combination method using the Cauchy distribution.
#'
#'
#'
#' @param weights a numeric vector/matrix of non-negative weights for the combined p-values. When it is NULL, the equal weights are used.
#' @param Pvals a numeric vector/matrix of p-values. When it is a matrix, each column of p-values is combined by ACAT.
#' @param is.check logical. Should the validity of \emph{Pvals} (and \emph{weights}) be checked? When the size of \emph{Pvals} is large and one knows \emph{Pvals} is valid, then the checking part can be skipped to save memory.
#' @return The p-value(s) of ACAT.
#' @author Yaowu Liu
#' @examples p.values<-c(2e-02,4e-04,0.2,0.1,0.8);ACAT(Pvals=p.values)
#' @examples ACAT(matrix(runif(1000),ncol=10))
#' @references Liu, Y., & Xie, J. (2019). Cauchy combination test: a powerful test with analytic p-value calculation
#' under arbitrary dependency structures. \emph{Journal of American Statistical Association},115(529), 393-402. (\href{https://amstat.tandfonline.com/doi/abs/10.1080/01621459.2018.1554485}{pub})
#' @export
ACAT<-function(Pvals,weights=NULL,is.check=TRUE){
Pvals<-as.matrix(Pvals)
if (is.check){
#### check if there is NA
if (sum(is.na(Pvals))>0){
stop("Cannot have NAs in the p-values!")
}
#### check if Pvals are between 0 and 1
if ((sum(Pvals<0)+sum(Pvals>1))>0){
stop("P-values must be between 0 and 1!")
}
#### check if there are pvals that are either exactly 0 or 1.
is.zero<-(colSums(Pvals==0)>=1)
is.one<-(colSums(Pvals==1)>=1)
if (sum((is.zero+is.one)==2)>0){
stop("Cannot have both 0 and 1 p-values in the same column!")
}
if (sum(is.zero)>0){
warning("There are p-values that are exactly 0!")
}
if (sum(is.one)>0){
warning("There are p-values that are exactly 1!")
}
}
#### Default: equal weights. If not, check the validity of the user supplied weights and standadize them.
if (is.null(weights)){
is.weights.null<-TRUE
}else{
is.weights.null<-FALSE
weights<-as.matrix(weights)
if (sum(dim(weights)!=dim(Pvals))>0){
stop("The dimensions of weights and Pvals must be the same!")
}else if (is.check & (sum(weights<0)>0)){
stop("All the weights must be nonnegative!")
}else{
w.sum<-colSums(weights)
if (sum(w.sum<=0)>0){
stop("At least one weight should be positive in each column!")
}else{
for (j in 1:ncol(weights)){
weights[,j]<-weights[,j]/w.sum[j]
}
}
}
}
#### check if there are very small non-zero p values and calcuate the cauchy statistics
is.small<-(Pvals<1e-15)
if (is.weights.null){
Pvals[!is.small]<-tan((0.5-Pvals[!is.small])*pi)
Pvals[is.small]<-1/Pvals[is.small]/pi
cct.stat<-colMeans(Pvals)
}else{
Pvals[!is.small]<-weights[!is.small]*tan((0.5-Pvals[!is.small])*pi)
Pvals[is.small]<-(weights[is.small]/Pvals[is.small])/pi
cct.stat<-colSums(Pvals)
}
#### return the ACAT p value(s).
pval<-pcauchy(cct.stat,lower.tail = F)
return(pval)
}
#'
#' A set-based test that uses ACAT to combine the variant-level p-values.
#'
#'
#' @param G a numeric matrix or dgCMatrix with each row as a different individual and each column as a separate gene/snp. Each genotype should be coded as 0, 1, 2.
#' @param obj an output object of the \code{\link{NULL_Model}} function.
#' @param weights.beta a numeric vector of parameters for the beta weights for the weighted kernels. If you want to use your own weights, please use the “weights” parameter. It will be ignored if “weights” parameter is not null.
#' @param weights a numeric vector of weights for the SNP p-values. When it is NULL, the beta weight with the “weights.beta” parameter is used.
#' @param mac.thresh a threshold of the minor allele count (MAC). The Burden test will be used to aggregate the SNPs with MAC less than this thrshold.
#' @return The p-value of ACAT-V.
#' @details The Burden test is first used to aggregate very rare variants with Minor Allele Count (MAC) < \emph{mac.thresh} (e.g., 10), and a Burden p-value is obtained. For each of the variants with MAC >= \emph{mac.thresh}, a variant-level p-value is calculated. Then, ACAT is used to combine the variant-level p-values and the Burden test p-value of very rare variants.
#'
#' If \emph{weights.beta} is used, then the weight for the Burden test p-value is demetermined by the average Minor Allele Frequency (MAF) of the variants with MAC < \emph{mac.thresh}; if the user-specified \emph{weights} is used, then the weight for the Burden test p-value is the average of \emph{weights} of the variants with MAC < \emph{mac.thresh}.
#'
#' Note that the \emph{weights} here are for the SNP p-vlaues. In SKAT, the weights are for the SNP score test statistics. To transfrom the SKAT weights to the \emph{weights} here, one can use the formula that \emph{weights} = (skat_weights)^2*MAF*(1-MAF).
#'
#' @author Yaowu Liu
#' @references Liu, Y., et al. (2019). ACAT: A fast and powerful p value combination
#' method for rare-variant analysis in sequencing studies.
#' \emph{American Journal of Humann Genetics 104}(3), 410-421.
#' (\href{https://www.sciencedirect.com/science/article/pii/S0002929719300023}{pub})
#' @examples library(Matrix)
#' @examples data(Geno)
#' @examples G<-Geno[,1:100] # Geno is a dgCMatrix of genotypes
#' @examples Y<-rnorm(nrow(G)); Z<-matrix(rnorm(nrow(G)*4),ncol=4)
#' @examples obj<-NULL_Model(Y,Z)
#' @examples ACAT_V(G,obj)
#' @export
ACAT_V<-function(G,obj,weights.beta=c(1,25),weights=NULL,mac.thresh=10){
### check weights
if (!is.null(weights) && length(weights)!=ncol(G)){
stop("The length of weights must equal to the number of variants!")
}
mac<-Matrix::colSums(G)
### remove SNPs with mac=0
if (sum(mac==0)>0){
G<-G[,mac>0,drop=FALSE]
weights<-weights[mac>0]
mac<-mac[mac>0]
if (length(mac)==0){
stop("The genotype matrix do not have non-zero element!")
}
}
### p and n
p<-length(mac)
n<-nrow(G)
###
if (sum(mac>mac.thresh)==0){ ## only Burden
pval<-Burden(G,obj, weights.beta = weights.beta, weights = weights)
}else if (sum(mac<=mac.thresh)==0){ ## only cauchy method
if (is.null(weights)){
MAF<-mac/(2*n)
W<-(dbeta(MAF,weights.beta[1],weights.beta[2])/dbeta(MAF,0.5,0.5))^2
}else{
W<-weights
}
Mpvals<-Get.marginal.pval(G,obj)
pval<-ACAT(Mpvals,W)
}else{ ## Burden + Cauchy method
is.very.rare<-mac<=mac.thresh
weights.sparse<-weights[is.very.rare]
weights.dense<-weights[!is.very.rare]
pval.dense<-Burden(G[,is.very.rare,drop=FALSE],obj, weights.beta = weights.beta, weights = weights.sparse)
Mpvals<-Get.marginal.pval(G[,!is.very.rare,drop=FALSE],obj)
Mpvals<-c(Mpvals,pval.dense)
if (is.null(weights)){
MAF<-mac/(2*n)
mafs<-c(MAF[!is.very.rare],mean(MAF[is.very.rare])) ## maf for p-values
W<-(dbeta(mafs,weights.beta[1],weights.beta[2])/dbeta(mafs,0.5,0.5))^2
}else{
W<-c(weights.dense,mean(weights.sparse))
}
is.keep<-rep(T,length(Mpvals))
is.keep[which(Mpvals==1)]<-F ## remove p-values of 1.
pval<-ACAT(Mpvals[is.keep],W[is.keep])
}
return(pval)
}
#'
#'
#' Get parameters and residuals from the NULL model
#'
#' Compute model parameters and residuals for ACAT-V
#'
#'
#' @param Y a numeric vector of outcome phenotypes.
#' @param Z a numeric matrix of covariates. Z must be full-rank. Do not include intercept in Z. The intercept will be added automatically.
#' @return This function returns an object that has model parameters and residuals of the NULL model of no association between genetic variables and outcome phenotypes. After obtaining it, please use \code{\link{ACAT_V}} function to conduct the association test.
#' @details \emph{Y} could only be continuous or binary. If \emph{Y} is continuous, a linear regression model is fitted. If \emph{Y} is binary, it must be coded as 0,1 and a logistic model is fitted.
#' @author Yaowu Liu
#' @examples Y<-rnorm(10000)
#' @examples Z<-matrix(rnorm(10000*4),ncol=4)
#' @examples obj<-NULL_Model(Y,Z)
#' @export
NULL_Model<-function(Y,Z=NULL){
n<-length(Y)
#### check the type of Y
if ((sum(Y==0)+sum(Y==1))==n){
out_type<-"D"
}else{
out_type<-"C"
}
#### Add intercept
Z.tilde<-cbind(rep(1,length(Y)),Z)
if (out_type=="C"){
#### estimate of sigma square
Z.med<-Z.tilde%*%solve(chol(t(Z.tilde)%*%Z.tilde)) ## Z.med%*%t(Z.med) is the projection matrix of Z.tilde
Y.res<-as.vector(Y-(Y%*%Z.med)%*%t(Z.med))
sigma2<-sum(Y.res^2)/(n-ncol(Z.med))
#### output
res<-list()
res[["out_type"]]<-out_type
res[["Z.med"]]<-Z.med
res[["Y.res"]]<-Y.res
res[["sigma2"]]<-sigma2
}else if (out_type=="D"){
#### fit null model
g<-glm(Y~0+Z.tilde,family = "binomial")
prob.est<-g[["fitted.values"]]
#### unstandarized residuals
Y.res<-(Y-prob.est)
### Sigma when rho=0
sigma2.Y<-prob.est*(1-prob.est) ### variance of each Y_i
### output
res<-list()
res[["out_type"]]<-out_type
res[["Z.tilde"]]<-Z.tilde
res[["Y.res"]]<-Y.res
res[["sigma2.Y"]]<-sigma2.Y
}
return(res)
}
Get.marginal.pval<-function(G,obj){
### check obj
if (names(obj)[1]!="out_type"){
stop("obj is not calculated from MOAT_NULL_MODEL!")
}else{
out_type<-obj[["out_type"]]
if (out_type=="C"){
if (!all.equal(names(obj)[2:length(obj)],c("Z.med","Y.res","sigma2"))){
stop("obj is not calculated from MOAT_NULL_MODEL!")
}else{
Z.med<-obj[["Z.med"]]
Y.res<-obj[["Y.res"]]
n<-length(Y.res)
SST<-obj[["sigma2"]]*(n-ncol(Z.med))
}
}else if (out_type=="D"){
if (!all.equal(names(obj)[2:length(obj)],c("Z.tilde","Y.res","sigma2.Y"))){
stop("obj is not calculated from MOAT_NULL_MODEL!")
}else{
Z.tilde<-obj[["Z.tilde"]]
Y.res<-obj[["Y.res"]]
sigma2.Y<-obj[["sigma2.Y"]]
n<-length(Y.res)
}
}
}
if (class(G)!="matrix" && class(G)!="dgCMatrix"){
stop("The class of G must be matrix or dgCMatrix!")
}
if (out_type=="C"){
G_tX.med<-as.matrix(Matrix::crossprod(Z.med,G))
### Sigma^2 of G
Sigma2.G<-Matrix::colSums(G^2)-Matrix::colSums(G_tX.med^2)
SSR<-as.vector((Y.res%*%G)^2/Sigma2.G)
SSR[Sigma2.G<=0]<-0
df.2<-n-1-ncol(Z.med)
t.stat<-suppressWarnings(sqrt(SSR/((SST-SSR)/df.2)))
marginal.pvals<-2*pt(t.stat,(n-1-ncol(Z.med)),lower.tail = FALSE)
}else if (out_type=="D"){
Z.stat0<-as.vector(Y.res%*%G)
### Sigma when rho=0
tG_X.tilde_sigma2<-as.matrix(Matrix::crossprod(G,Z.tilde*sigma2.Y))
Sigma2.G<-Matrix::colSums(G^2*sigma2.Y)-diag(tG_X.tilde_sigma2%*%solve(t(Z.tilde)%*%(Z.tilde*sigma2.Y))%*%t(tG_X.tilde_sigma2))
marginal.pvals<-2*pnorm(abs(Z.stat0)/sqrt(Sigma2.G),lower.tail = FALSE)
}
return(marginal.pvals)
}
Burden<-function(G,obj,kernel="linear.weighted",weights.beta=c(1,25),weights=NULL){
### check obj
if (names(obj)[1]!="out_type"){
stop("obj is not calculated from NULL_MODEL!")
}else{
out_type<-obj[["out_type"]]
if (out_type=="C"){
if (!all.equal(names(obj)[2:length(obj)],c("Z.med","Y.res","sigma2"))){
stop("obj is not calculated from NULL_MODEL!")
}else{
Z.med<-obj[["Z.med"]]
Y.res<-obj[["Y.res"]]/sqrt(obj[["sigma2"]]) ## rescaled residules
n<-length(Y.res)
}
}else if (out_type=="D"){
if (!all.equal(names(obj)[2:length(obj)],c("Z.tilde","Y.res","sigma2.Y"))){
stop("obj is not calculated from NULL_MODEL!")
}else{
Z.tilde<-obj[["Z.tilde"]]
Y.res<-obj[["Y.res"]]
sigma2.Y<-obj[["sigma2.Y"]]
n<-length(Y.res)
}
}
}
### MAF
MAF<-Matrix::colSums(G)/(2*dim(G)[1])
p<-length(MAF)
#### weights
if (kernel=="linear.weighted"){
if (is.null(weights)){
W<-dbeta(MAF,weights.beta[1],weights.beta[2])
}else{
if (length(weights)==p){
W<-weights
}else{
stop("The length of weights must equal to the number of variants!")
}
}
}else if (kernel=="linear"){
W<-rep(1,p)
}else{
stop("The kernel name is not valid!")
}
###### if G is sparse or not
if (class(G)=="matrix" || class(G)=="dgCMatrix"){
if (out_type=="C"){
Z.stat.sum<-as.vector((Y.res%*%G)%*%W)
Gw<-G%*%W
sigma.z<-sqrt(sum(Gw^2)-sum((t(Z.med)%*%(Gw))^2))
}else if (out_type=="D"){
Z.stat.sum<-as.vector((Y.res%*%G)%*%W)
Gw<-as.vector(G%*%W)
sigma.z<-sum(Gw^2*sigma2.Y)-((Gw*sigma2.Y)%*%Z.tilde)%*%solve(t(Z.tilde)%*%(Z.tilde*sigma2.Y))%*%t((Gw*sigma2.Y)%*%Z.tilde)
sigma.z<-as.vector(sqrt(sigma.z))
}
}else{
stop("The class of G must be matrix or dgCMatrix!")
}
V<-Z.stat.sum/sigma.z
Q<-V^2 ## Q test statistic
pval<-1-pchisq(Q,df=1)
return(pval)
}
Maintained by Dylan Lawless. Scientist at EPFL. Resume and Google scholar. RSS feed