Pharmacogenomics for personal medicine
- Introduction
- Download VCF data
- Annotate VCF data
- Annotating a full VCF
- Comparing annotated genetic data to drug lists
- Full-scale merging genetic and pharmacogenomic data
- Downstream interpretation
- Drug indication
- Understanding variant annotation
- Unknown variants
- Gene dosage
- Drug indication
- A large scale example summary
- Funding strategy
- Legal requirements
- Cytochrome P450 (CYP) genes for known PGx
- References
Introduction
With the popularisation of commercial genetics services, more and more people are now able to “decode” their genetic data. Questions that might arise from this information include “do I have potentially disease-causing variants that can be treated with a drug?”, or “am I taking a drug that will be affected by my genetics?”. To tackle such questions with an example, we use public data in combination with pharmacogenomics.
The most common file type for storing DNA variant data is the VCF format: What is a vcf and how should I interpret it?.
Download VCF data
Example VCF from https://my.pgp-hms.org/public_genetic_data:
- A randomly selected whole genome VCF file:
- https://my.pgp-hms.org/profile/hu24385B:
- filename: hu24385B 2019-04-07.vcf.gz
- sequence provider: Dante Labs Whole Genome
- size: 147 MB
- Additional examples - not tested:
- [1] VCF from Dante Labs vs GRCh37 (246 MB) https://my.pgp-hms.org/profile/hu1D5A29
- [2] WGS 30x filtered SNP VCF (325 MB) https://my.pgp-hms.org/profile/hu1C1368
- [3] 60820188474283.snp.vcf.gz (222 MB) https://my.pgp-hms.org/profile/hu6ABACE
The “hu24385B” VCF has 3,461,639 variants. VCF files can contain a large range of information for each variant, however only the first 7 column are strictly neccessary; Chromosome, position, ID, Reference, Alternate, Qulaity, Filter, info. The details are explained on this GATK forum post.
Annotate VCF data
Annotation information about the gene name (or related diseases) is often not present when the VCF is generated and only added later. To get the gene names, the simplest way was is to upload a VCF (or a part of it) to Variant Effect Predictor. This will supply the gene symbol (and any other information about each DNA variant).
To reduce the time and output you can limit the options:
- Split the file and run in batches to save time.
- Test the first ~1800 variants:
head -2000 56001801068861_WGZ.snp.vcf > test.vcf
-
Then annotate with Variant Effect Predictor.
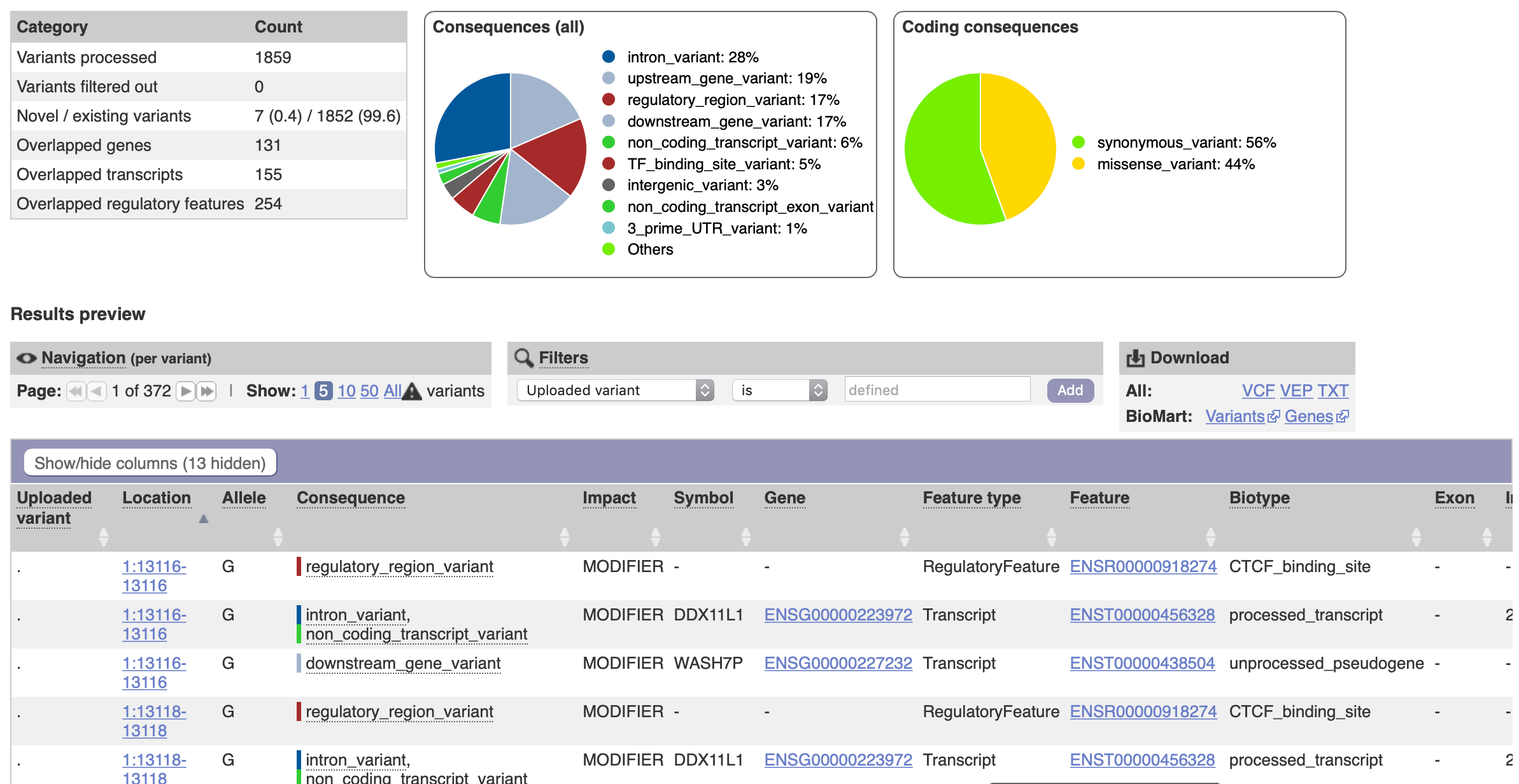
- Make certain that you use the same reference genome as used on the input data.
- The VCF file was made using reference genome GRCh37.
- Therefore, the Ensembl/VEP website URL should be for that genome build (grch37, not the default GRCh38).
Annotating a full VCF
- One could annotate a whole genome using the Ensembl web interface.
- However, one would need to split your VCF into smaller block first.
- For routine usage the command-line version of VEP and it’s databases should be installed on run locally.
- I will provide a completed annotated VCF for you.
There are several bioinformatics tools that are commonly used for manipulating genetic file formats such as VCFtools. However, to get a real understanding of the data type, here is a method using command line bash to split a VCF file into smaller blocks. A bash script is printed below where I use very mainstream traditional command-line tools to wrangle data, including gunzip to unzip compressed files, wc to count lines, cat to print a file, head to read the top of a file, sed to edit lines, awk for data extraction, and grep for text search (not used).
Putting the code below into a file with the filename extension with “.sh” will allow it to be run by your terminal, in this case using the bash language. I encourage you to read each line and figure out what should happen. If it makes sense then it is reasonable to swap such a manual method with a more efficient specialised tool.
#!/bin/bash
# VEP accept files of <50MB size.
# We will split our large VCF into smaller files.
# Each file requires the same original
# headers and file extension ".vcf"
# Unzip the VCF.gz
gunzip 56001801068861_WGZ.snp.vcf.gz
# Count the number of lines in vcf
wc -l 56001801068861_WGZ.snp.vcf
# How should a vcf file look?
# See the links posted in this tutorial above
# Take a look at the header
# This VCF has 140 lines of header metadata (beginning with "#")
# Line 141 shows the column headers: CHROM POS ID REF ALT...
# Line 142 starts with the first variant
head -142 56001801068861_WGZ.snp.vcf
# Print the header to a new file for later
head -141 56001801068861_WGZ.snp.vcf > header
# Print everything else (the body) to
# a new file that we will then split.
sed '1,141d' 56001801068861_WGZ.snp.vcf > body.vcf
# Make a new directory for the next step
mkdir split_files
# Move the large file inside
mv body.vcf split_files/
cd split_files
# Now split the body.vcf into smaller
# files of 200,000 lines each
split -l 150000 body.vcf
# You will now how ~10 files "xaa, xab, etc."
# Add the header back onto all of these files to make them VCFs again.
# This "for loop" will do the following for each file:
# Print the header and the vcf body to
# a file with the same name,
# adding a file extension ".vcf".
# Then remove the vcf body file that
# does not have the ".vcf" extension
# leaving you with the original whole genome VCF split
# into smaller files, each with the same headers.
for file in ./x* ;
do cat ../header $file >> $file.vcf && rm $file ;
done
# These should be small enough to run on VEP online.
# You could edit the split command to make a
# reasonable number of files,
# uploading >10 is not efficient.
Comparing annotated genetic data to drug lists
- We now assume that we have a VCF where VEP annotated each variant.
- The features can include gene names, variant consequence, pahtnogenicy prediction, etc.
- The next aim is to see if any output genes are also present in your drug-gene database.
- The method will require merging both dataset (gene and drug datasets) based on shared features.
- A simple sanity test would be useful first.
Drug-gene list
Here is a drug-gene list “similar” to the private version from DrugBank:
Drugbank has changed their access policy and now requires access applications. If you want the up-to-date DrugBank data I believe you can apply for non-commercial use (and wait a few days). The data can then be found at: https://go.drugbank.com/releases/latest#protein-identifiers, “protein-identifiers” tab, “Drug Target Identifiers” section, “All” file.
Example of how to check gene list versus drug-gene list
- Extract the gene symbols column from VEP output
- Compare gene symbols to a list of druggable target genes from DrugBank.
# Get a list of unique gene symbols
cut -f1 -d "," vep_output_file.csv - uniq > unique.genes.txt
# - Cut column 1 (f1)
# - with a delimiter comma (,)
# - from the vep output csv file (or tsv, or text file)
# - then pipe (\|) that result into another program (sort) to sort the result in alphabetic order
# - pipe (\|) again this result into(uniq)
# - so that only one unique gene name is output
# - then (>) write the output into the new file "unique.genes.txt".
- Repeat the same method on the DrugBank dataset
- Output the gene names from DrugBank to “unique.druggable.txt”
# Get a list of gene symbols which are present in both datasets
sort unique.genes.txt unique.druggable.txt - uniq -c -i | grep -v '1 '
This command also used “uniq -c” to count how many times each gen name occurs and then “grep -v ‘1 ‘” meaning ignore genes that are only present 1 time. We want the genes that are present twice, once in each list.
- The genes which were in present in both the variant list and DrugBank list are:
- From a 2,000 line VCF file:
- From a 10,000 VCF line file:
Full-scale merging genetic and pharmacogenomic data
The following R language script is used to merge the VEP annotated VCF file with a DrugBank database based on the gene names that are common to both datasets. Read each line and try to understand the process. I recommending installing R and then installing R studio to edit and run your commands.
# Comment lines are ignored because of "#" symbol.
# Command lines are run by clicking "Run" or "command+enter" on Mac
# csv = comma sep file
# tsv = tab spaced
# txt = white space
#///////////////////////////////
# To do
#///////////////////////////////
# Files: vepfile, drugs
# merge based on Gene symbol
#///////////////////////////////
# import the VEP file
#///////////////////////////////
# Important note:
# The VEP file will start with a header line
# that begins with "#" symbol. But this is being ignored by R.
# Open the file with text edit and remove that symbol.
# Maybe there is an R command to do this on import, whatever is faster.
# Split vcf into ~ 10 files.
# file 1 "xaa" was anylised on VEP,
# download the TXT version,
# or unzip my provided example one - cfKJCLRm0eKXsaEG.txt.zip
# https://grch37.ensembl.org/Homo_sapiens/Tools/VEP/
# Import the VEP output
vepfile <- read.table(
file="cfKJCLRm0eKXsaEG.txt",
na.strings=c("", "NA"),
sep="\t",
header=TRUE)
# Import drugbank table
# the "fill=TRUE" is needed because not all
# file lines have the same number of elements.
drugs <- read.table(
file="drugbank_all_interaction.txt",
na.strings=c("", "NA"),
sep="\t",
header=TRUE,
fill = TRUE )
# We can merge these two files based on 1 common column.
# However, the gene name column does not have the same name.
# One of them needs to be renamed:
# vepfile="SYMBOL"
# drugs="Gene.Name"
# colnames(df)[colnames(df) == 'oldName'] <- 'newName'
colnames(vepfile)[colnames(vepfile) == "SYMBOL"] <- "Gene.Name"
# Merge keeping only matches
merged <- merge(x = vepfile,
y = drugs,
by = "Gene.Name", all = FALSE)
# Remove empty data "NA"
# Install packeges once (comment out then)
# Load library each time to use "%>%" (command join) and filtering
install.packages("tidyr")
library(tidyr)
install.packages("dplyr")
library(dplyr)
df1 <- merged %>% drop_na(Drug.IDs)
# Make a list of benign variant types that should be removed
filter_out <-
'synonymous_variant|UTR|NMD_transcript\
|non_coding|downstream|upstream\
|intron|mature_miRNA_variant'
# Then filter out anything matching these terms.
df2 <- df1 %>% filter_all(all_vars(!grepl(filter_out,.)))
# Save an output tsv file for Excel, etc.
write.table((df2), file='./output.tsv', sep="\t", \
quote=FALSE, row.names=FALSE)
If you complete this process the output will contain a perfectly merged dataset.
Downstream interpretation
The next challenge lies downstream in interpreting which variants can have an effect that would justify the use of the drug.
- Are non-coding or synonymous variants worth reporting?
- Genes have multiple transcripts which means one variant can be both coding and non-coding depending on transcript splicing, etc.
- How can we integrate pharmacodynamics, covariates to drug response, contraindications, variant pathogenicity, etc.
Drug indication
My example used DrugBank for pharmacogenomic information. Another option is to use the FDA information as the primary source.
Is the gene-drug interaction good or bad?
Drugs might be either a treatment for a genetic determinant, or a warning for drug usage in someone who also has a genetic variation that might effect their treatment. The “Labelling Section” listed by FDA might offer the best information.
https://www.fda.gov/drugs/ For example, if we go and check the Prescribing Information PDF to compare two drugs we see that
(1) One is used to directly block a gene product,
(2) The second warns about use with certain genetic complications.
Drug 1: Atezolizumab (1),
Gene: CD274 (PD-L1)
Labeling: Indications and Usage
PRESCRIBING INFORMATION: TECENTRIQ (Atezolizumab) is a programmed death-ligand 1 (PD-L1) blocking antibody indicated for the treatment of patients with…
linked PDF.
Explained: Genetic disorder and the drug to treat it, exactly what you want.
Drug 2: Avatrombopag (3)
Gene: PROC
Labeling: Warnings and Precautions
PRESCRIBING INFORMATION: Thrombotic/Thromboembolic Complications: DOPTELET is a thrombopoietin (TPO) receptor agonist… Monitor platelet counts and for thromboembolic events
linked PDF.
Explained: Atezolizumab is used to treat thrombocytopenia (low levels of thrombocytes).
You do not want to give this to someone who has PROC deficiency;
their disease is Thrombophilia (hypercoagulability, or thrombosis).
With this in mind, perhaps an application doing this job could work two ways.
(1) If someone has a genetic disorder, the drug, gene, and Indicated usage appears.
(2) If someone is prescribed a drug a suggestion appears to check their genetics, with a link to the gene and Warnings and Precautions.
Understanding variant annotation
Variant Effect Predictor (VEP) is very useful.
During variant annotation, VEP supplies a “consequence” column.
Consequences are general and based on translation of genetic code in humans.
The Loss-of-function (LoF) consequence is the simplest example (splice, stop mutations).
The variant consequence may be one of the defining criteria by which variants can
be included in analysis since they are interpretable or of ostensibly known significance.
Note: Using this alone could introduce spurious results so it is best to have a solid criteria
for selecting consequences of interest.
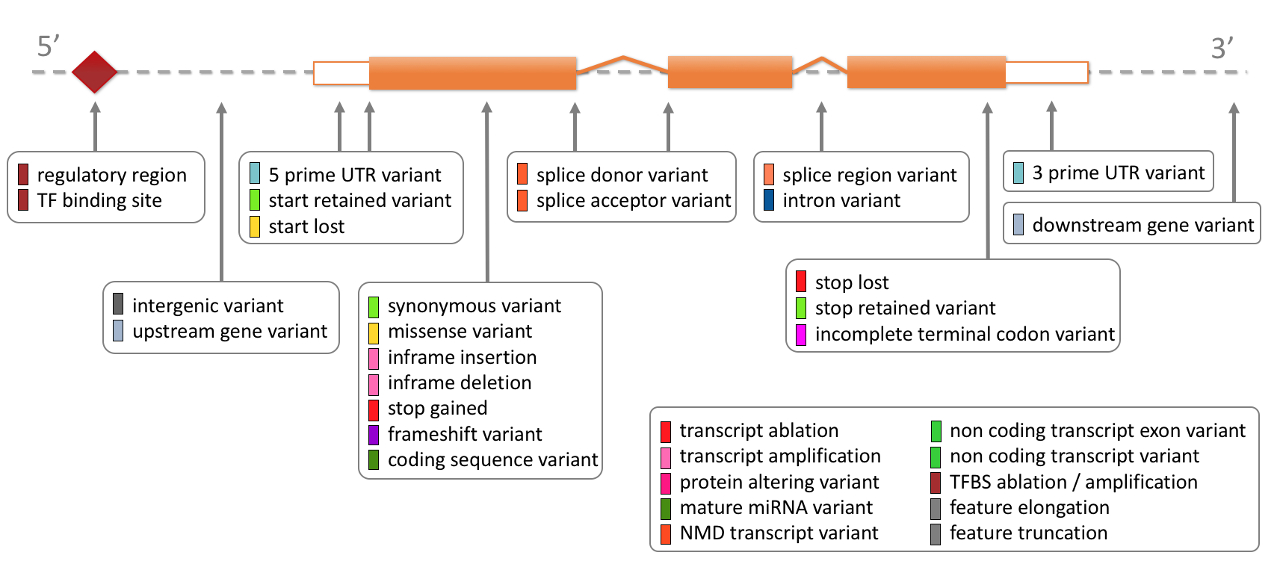
The consequences provided by VEP are too long to discuss in detail here.
The table from the ensembl website is worth reading; the HIGH impact variants
might be a simple method for selecting candidates:
Ensembl Variation - Calculated variant consequences.
Note: For a real product, the code should be run offline (a perl program with a few local library dependencies). The databases/cache that it uses are a bit too large to include on in a user software. In the real world you would have to send anonymised packets from the user via an API for accessing the genomic databases hosted on your servers. Make sure to check their license to see if you can use oftware and databases in a commercial product. http://www.ensembl.org/info/about/legal/code_licence.html
Running the software:
- Using VEP is a vital part of converting the DNA variant information (genome position and nucleotide change) into annotated variant effects (protein coding change, gene name, predicted pathogenicity).
- It requires the VEP code to run and requires a copy of the database files (reference genome, gene information, etc.).
- You can upload a small number of variants to the online VEP web server to do this, or you can download the database and code to run on your own computer/server.
So to process your customer/patient data, you have to choose one of these methods:
- Customer must upload their entire file to your server that runs VEP (1GB - 30GB per individual genome data).
- Customer must download the database and VEP code to run on their own computer (complex and large download for them, not recommended).
Number 1 is better. But sending one large file often has problems. If they have a VCF file, you could:
- Break it into small equal sized blocks to upload the data to you. Anonymisation is normal for all internet connections now, but you could just mention that some method of anonymising these blocks is important since someone “hacker” might try to steal information if most of the network data being sent to you contains small VCF format files.
- You could also run a very small piece of code on the cusotmers software application that could extract just the main parts of the VCF file that you need, instead of sending everything. This is explained in sections 7-9. You could say this, but don’t need to actually have working. i.e. the drugbank information only includes a certain number of human genes so perhaps you could just extract these using a list of genome coordinates before processing with VEP.
For the license:
- Anyone is free to download and use VEP code.
- However, if you modify or reuse the code commercially it might affect the possibility of getting a patent for your product.
- Your product uses VEP as an intermediate step, so you probably only need to include credit, or other legal info to say you have used it.
- If there were a reason to prevent you using the software commercially, you might be able to make a simple replacement that could give the minimum outputs that you need - gene name and mutation type. If the topic happens to interest you, you can read about reverse engineering software.
- As an aside, you could also decide that you don’t want to commercialise and offer this tool for free which would prevent bigger companies (like Google) from offering this service in return for harvesting the public’s genetic data.
Optimising VCF annotation
The slowest part of the method is VCF annotation. You can significantly increase the speed by first reducing the input to contain only regions of interst. That is, prepare a list of coordinates for each gene, and select for those regions in your input VCF or genotype data before annotation (VEP).
How to get coordinates for a gene list
Use Biomart.
Their main server was down when I tried, so I went via Ensembl, data access section:
http://www.ensembl.org/info/data/biomart/index.html
Then to use the BioMart data mining tool
http://www.ensembl.org/biomart/martview/
I actually needed the positions using GRCh37 (rather than 38), so I switched to the old Ensembl using
http://www.ensembl.org/info/website/tutorials/grch37.html
to get to http://grch37.ensembl.org/index.html
then the Biomart section
http://grch37.ensembl.org/biomart/martview/
Choose DataBase: Genes 93 Dataset: Human Filter -> Gene -> Input external ref ID list -> (change dropdown) Gene
Name paste your list.
e.g. VPS45 PSMB8 BLNK NEFL NLRP7 SMAD4 PSMB9
To set the output type: Attributes -> Gene -> select “gene start”, “gene stop”, “gene name”, or anything extra.
Select the “Results” button at the top and export.
The results can be tsv or csv.
You would have to figure out how to extract the regions from the vcf (sed, grep, awk, R code, etc.).
When I needed this, I used my own tools which required converting to format like this “X:1-2000”, and ordered by number and alphabetic (some positions in the reference genome were patches added later and have an alphanumeric instead of the normal chromosome).
If you use this list to extract regions from a VCF, remember to include all the original VCF header information.
Extracting regions from a VCF using a bed file
The early part of this tutorial shows how old-school command line tools can be used to extract data.
Indeed, this may be computationally most efficient but there are some specialised tools that make the process easier in general.
You can use VCFtools to extract specified regions.
https://vcftools.github.io/man_latest.html
You could use a list of defined genome position to reduce the size of your dataset.
The defined genomic coordinates are generally supplied in a file format called the (BED file](https://en.wikipedia.org/wiki/BED_(file_format))
Note that sometimes the bed file “chrom” ID - the name of the chromosome (e.g. chr3) does not match if the VCF file uses “3” instead of “chr3”.
You might need to edit the bed.
My bed file was like this:
(tab spaced), ref.bed
chrom chromStart chromEnd
1 3549 13555
And this command ran OK for me to give “output_prefix.recode.vcf”
$:~/tools/vcftools_0.1.13/bin \
./vcftools \
--vcf ~/input.vcf \
--bed ~/ref.bed \
--out output_prefix \
--recode --keep-INFO-all
This new VCF will now only contain gene regions that are potentially “druggable”, or at least included on the FDA list. VCF annotation will be much faster than annotation of the whole genome.
Unknown variants
In the majority of situations you will be stuck with variants of unknown significance.
In the absence of tailored analysis and interpretation of each invidual variant, one must often rank unknown variants based on a predicted pathogenicity.
Carefully consider that predictions can be completed wrong and address how such an annotation should be presented.
One can rank unknown variants based on PHRED-scaled CADD score, highest being more predicted pathogenic.
https://cadd.gs.washington.edu/info
Polyphen gives a predicted outcome label and a probability score 0-1 from benign to probably damaging.
See what other pathogenicity prediction tools you can find and estimate how widespread/accepted their usage is.
Gene dosage
An important cosideration of variant effect depends on gene dosage. A dominant gene may be affected by a single heterozgous variant while a recessive gene may be able to compensate against the negative effect of a heterozyous variant due the presence of a second functional gene copy. Therefore, the presence of heterozygous or homozygous allele is an important consideration. Some genes may be sensitive to a hemizygous effect, low frequency somatic variants, mosaisism, etc. SNV calling in NGS is a broad topic, but it is safe to say that at least the allele dosage (generally heterozygous or homozygous) should be included in result summary. If possible, when a gene is linked to a specific disease then the inheritance type associated with that gene-disease should also be included. For example, https://www.omim.org is a good place to see examles. The genetic disease cystic fibrosis is shown with an inheritance type AR (autosomal recessive) meaning that damaging variants on both gene alleles are required to cause disease. The gene cftr, which is the genetic determinant of cystic fibrosis, also has an OMIM page cftr that also lists AR inheritance. An excellent resource for matching gene to disease is the https://panelapp.genomicsengland.co.uk. Individual genes can be explored, or “panels” of disease-specific gene lists can be explored. For example, here is the “Bleeding and platelet disorders” panel. This shows the “Mode of inheritance” and colour-coded confidence in the disease-gene relationship. Integrating this type of expert-curated open datasets can be extremely useful.
Drug indication
The indication or warning can be difficult to automate.
For the example drug
https://www.drugbank.ca/drugs/DB11595
the section “Pharmacology” “Indication” has the Indication info.
The FDA label is contained as a PDF attachment in the section “REFERENCES” FDA label Download (245 KB).
If I had to automate the process I would add a URL link for each drug:
for gene name CD274
the drugbank column Drug IDs has these:
DB11595; DB11714; DB11945
and for each ID you could append the ID onto the drugbank URL to link to the webpage
https://www.drugbank.ca/drugs/.
You can do this in R with some technical how-to reading, or do it manually for a quick example like this and removing space to create a web URL.
URL Drug IDs
https://www.drugbank.ca/drugs/ DB00303
https://www.drugbank.ca/drugs/ DB00114
https://www.drugbank.ca/drugs/ DB00142
https://www.drugbank.ca/drugs/ DB01839
https://www.drugbank.ca/drugs/ DB00125
A large scale example summary
I do not suggest this for a small project, but if I was to automate subsection requests for real:
- [1] Download the whole database (probably a big table sized >100MB) and
- [2] For every query (the Drug ID) extract the sections of interest (indication, Biologic Classification, Description, FDA label, etc.)
- [3] Display each section as additional columns in candidate genes table.
- [1] Would be here: https://www.drugbank.ca/releases/latest
- [2] Would be like this: https://www.w3schools.com/xml/default.asp
Look at example 2, your database request might be something like: [get food name = Belgian Waffles, description] or [get drug ID = DB11595, indication, Biologic Classification, Description, FDA label.] The database request problem can be tricky to optimise but not especially difficult with some experience in SQL-type management. - [3] For every line in the gene candidate table, do this query request and output the result into the same row.
The final table would be something that includes colunm headers like:
Gene, consequence, variant, amino acid, genome position, CADD, DrugBank ID, Description, Indication, FDA label PDF link, etc. This table could be ranked based on consequence, CADD score. The top couple of rows then might be converted into a more readable format like a PDF.
Funding strategy
University-based start-ups ususally follow a plan with three or four funding stages before coming to market. It is also possible to get investors from day 1, but it is more usual to follow the steps outlined here. It is also possible to partner with early investors for their guidance rather than for financial investment.
Example funding stages:
- A - Fundamental research (e.g. SNSF)
- B - Tech development (gap between the basic research and usable product)
- C - Product development (e.g. industry, Innosuisse funding)
Example funding sources per stage:
- [A] Ignite grant
- https://www.epfl.ch/innovation/startup/grants/ignition-grants/
- 30K - 6 month, salary/consumables
- [A] Innogrant
- <https://www.epfl.ch/innovation/startup/grants/innogrants/
- 100K - 1 year, salary for startup founder.
- [B] BRIDGE Proof of concept
- https://www.bridge.ch/en/proof-of-concept/
- 130K - 1 year
- [B] BRIDGE Discovery
- https://www.bridge.ch/en/discovery/
- (alternative to Proof of concept, more for experienced researchers).
- [C] Innosuisse
- Federal funding for startups of social benefit, etc.
- https://www.innosuisse.ch/inno/en/home/start-and-grow-your-business/startup-coaching.html
- [C] Other investors, venture capital, or investing from big companies like J&J, Pfizer, &c. For example, in a recent J&J meeting the start-up-related experts discussed how they work with startups.
https://advancesindrugdiscovery.splashthat.com Basically, the “innovation” department experts help you to figure out what stage you are at. You can contact them as soon as you can disclose your tech non-confidentially (either you have patent protection or do not need it). If you were taking this path you would probably have competed steps 1-4.
Legal requirements
Swiss law
I include both the English and French translations here as the original source does not always include full English translation. Swiss law contains specific provisions on genetic testing in humans. The Federal Council is divided into 7 departments and one chancellory. Each department contains their relevant offices (usually fewer than 10). For our interests, the governing hierarchy order is as follows:
- Le Conseil fédéral
- The Federal Council
- Département fédéral de l’intérieur (DFI),
- Federal Department of Home Affair (FDHI),
- Office fédéral de la santé publique (OFSP).
- Federal Office of Public Health (FOPH).
This office is then responsible for their relevant ordinances as organised under internal law: https://www.fedlex.admin.ch/en/cc/internal-law/8. A direct weblink to our area of interest is available on Législation Analyses génétiques, but it is useful to view the legal framework in context instead of abstractly.
- Internal law (1-9 sec)
- Droite interne (1-9 sec)
- Sec 8..: (81-86 subsections) Health - Employment - Social security
- Sec 8..: (81-86 subsections) Santé - Travail - Sécurité sociale
- Sec 81.: Health: (810-819 subsubsections)
- Sec 81.: Santé: (810-819 subsubsections)
- Sec 810: Medicine and human dignity
- Sec 810: Médecine et dignité humaine
- Sec 810.1: Medically assisted reproduction and genetic engineering in the human field
- Sec 810.1: Procréation médicalement assistée et génie génétique dans le domaine humain
- Sec 810.12: Federal Act of 8 October 2004 on Human Genetic Testing (HGTA)
- Sec 810.12: Loi fédérale du 8 octobre 2004 sur l’analyse génétique humaine (LAGH)
This contains section (810.12) contains 10 sections with 44 articles covering the initial regulations.
Three ordinance then include further details (with several sections, articles, or annexes each):
- Sec 810.122.1: Ordinance of 14 February 2007 on Human Genetic Analysis (OAGH)
- Sec 810.122.1: Ordonnance du 14 février 2007 sur l’analyse génétique humaine (OAGH)
- Sec 810.122.122: Ordinance of the Federal Department of Home Affairs of February 14, 2007 on Human Genetic Analysis (OAGH-DFI)
- Sec 810.122.122: Ordonnance du DFI du 14 février 2007 sur l’analyse génétique humaine (OAGH-DFI)
- Sec 810.122.2: Ordinance of February 14, 2007 on DNA profiling in civil and administrative matters (OACA)
- Sec 810.122.2: Ordonnance du 14 février 2007 sur l’établissement de profils d’ADN en matière civile et administrative (OACA)
The details are then listed individually at: https://www.fedlex.admin.ch/fr/cc/internal-law/81#810.12 and as stated, includes authorisation of “Pharmacogenetic tests performed to determine the effects of a planned therapy”, “analyses pharmacogénétiques effectuées dans le but de déterminer les effets d’une thérapie prévue”.
Accreditation
ISO 15189 is a commonly sought standard accreditation for genetic analysis labs, which is carried out by recognized accreditation services like FINAS. Here it is mentioned for the Geneva health 2030 genome center for clinical grade sequencing: https://www.health2030genome.ch/dna-sequencing-platform/. Other additional ISO accreditation standard concern Genomic information representation, including 23092-4 Reference software or 23092 Transport and storage of genomic information.
GA4GH provides other information about many legal and ethic topics. BlueprintGenomics is a good example company for comparison: https://blueprintgenetics.com/certifications/.
Cytochrome P450 (CYP) genes for known PGx
Cytochrome P450 monooxygenases are a group of genes encoding proteins that catalyze the oxidation and metabolism of a large number of xenobiotics and endogenous compounds. Therefore these genes/proteins are important for drug metabolism. Furthermore, common genetic variants are known for many of these genes which affect how the protein interacts with drugs. The Pharmacogene Variation (PharmVar) consortium repository is used to label human cytochrome P450 (CYP) genes for known PGx variation. The major focus of PharmVar is to catalogue allelic variation of genes impacting drug metabolism, disposition and response and provide a unifying designation system (nomenclature) for the global pharmacogenetic/genomic community. Similar resource include the Pharmacogenomic KnowledgeBase, and the Clinical Pharmacogenetic Implementation Consortium. You can read more in this post on one example of pharmacogenomic analysis using these resources.
References
- https://www.fda.gov/drugs/science-research-drugs/
- https://www.pharmgkb.org/view/drug-labels.do
- Mary V. Relling & William E. Evans. Pharmacogenomics in the clinic. Nature 2015; 526, 343–350. doi: 10.1038/nature15817
- Yip VL, Hawcutt DB, Pirmohamed M. Pharmacogenetic Markers of Drug Efficacy and Toxicity. Clin Pharmacol Ther. 2015;98(1):61-70. doi: 10.1002/cpt.135.
- David R. Adams, M.D., Ph.D., and Christine M. Eng, M.D. Next-Generation Sequencing to Diagnose Suspected Genetic Disorders N Engl J Med Oct 2018 doi: 10.1056/NEJMra1711801
Maintained by Dylan Lawless. Scientist at EPFL. Resume and Google scholar. RSS feed