Pharmacogenomics from a VCF file
- Audience
- What pharmacogenomics asks
- What this workflow produces
- Input files
- Resources used
- Example VCF
- Inspect the VCF
- Check the reference genome
- Annotate variants with VEP
- Split a VCF for web annotation
- Run VEP locally
- Clean the VEP output
- Build a small pharmacogene list
- Compare VCF genes with pharmacogenes
- Filter VEP rows to pharmacogenes
- Reduce the VCF before annotation
- Prepare a drug-gene table
- Merge VEP and drug-gene data in R
- Merge VEP and drug-gene data in Python
- Read the candidate table
- Consequence is not interpretation
- Known pharmacogenomic alleles
- Zygosity and dosage
- Drug-gene relationship types
- Unknown variants
- Limits of the first-principles workflow
- Practical summary
- References and resources
Audience
This page is written for a postgraduate class on precision medicine.
The aim is to understand how a pharmacogenomics analysis works under the hood. Professional laboratories and clinical systems usually use validated software, curated databases, quality-controlled pipelines, and formal reporting rules. Those systems are necessary for real use.
The exercise here is different. We start with a VCF file and build the logic ourselves. The point is to see the file types, the columns, the joins, the assumptions, and the failure points. Once those are clear, packages and pipelines become easier to judge.
What pharmacogenomics asks
Pharmacogenomics studies how genetic variation affects drug metabolism, toxicity, dosing, and response.
A pharmacogenomic result usually connects five things:
| Component | Example |
|---|---|
| Gene | CYP2C19 |
| Allele or genotype | CYP2C192/17 |
| Drug | Clopidogrel |
| Phenotype | Intermediate metaboliser |
| Recommendation source | CPIC or DPWG guideline |
A variant in a drug-related gene is not automatically pharmacogenomic evidence. A drug may bind a protein, a gene may encode a metabolising enzyme, or a variant may be present in a pharmacogene. These are different facts.
This distinction matters throughout the analysis.
What this workflow produces
The workflow produces a candidate table.
VCF
↓
variant annotation
↓
gene and consequence extraction
↓
pharmacogene matching
↓
drug-gene annotation
↓
candidate table
The candidate table can show that a person has variants in genes connected to drugs. It does not, by itself, provide a clinical pharmacogenomic report.
A clinical report requires validated allele calling, diplotype interpretation, quality control, guideline mapping, and clinical review.
Input files
The exercise uses five file types.
| File | Purpose |
|---|---|
input.vcf.gz |
Genetic variants from one genome or exome |
annotated_vep.tsv |
VEP annotation table |
pharmacogenes.txt |
Genes of interest |
drug_gene_table.tsv |
Drug-gene relationships |
candidate_output.tsv |
Merged candidate table |
A real workflow may also use BED files, reference FASTA files, VEP cache files, gene panels, PharmVar allele definitions, CPIC tables, and FDA drug labels.
Resources used
Each resource answers a different part of the pharmacogenomic question.
| Resource | Role |
|---|---|
| Ensembl VEP | Converts genomic coordinates into gene and transcript annotations |
| MANE Select | Defines preferred matched Ensembl and RefSeq transcripts |
| PharmVar | Defines star alleles for pharmacogenes |
| CPIC | Provides clinical pharmacogenetic implementation guidelines |
| PharmGKB | Curates drug-gene evidence, annotations, labels, and pathways |
| DPWG | Provides pharmacogenetic dosing recommendations |
| FDA pharmacogenomic biomarker table | Links drug labels to pharmacogenomic biomarkers |
| DrugBank | Lists drug targets, enzymes, carriers, transporters, and drug metadata |
| ClinVar | Curated variant-level clinical assertions |
| gnomAD | Population allele frequencies |
DrugBank is useful for drug-target relationships. CPIC, PharmGKB, PharmVar, and DPWG are closer to clinical pharmacogenomic interpretation.
Example VCF
Public example genomes are available from the Personal Genome Project.
The original classroom exercise used:
Profile: hu24385B
Source: Personal Genome Project
File: hu24385B 2019-04-07.vcf.gz
Sequencing provider: Dante Labs Whole Genome
Approximate size: 147 MB
Approximate variants: 3.46 million
A VCF is a structured text file. It contains metadata, a column header, and one row per variant.
| Column | Meaning |
|---|---|
| CHROM | Chromosome or contig |
| POS | Position on the reference genome |
| ID | Variant identifier, often rsID |
| REF | Reference allele |
| ALT | Alternate allele |
| QUAL | Variant quality score |
| FILTER | Pass or filter status |
| INFO | Variant-level annotations |
| FORMAT | Genotype field definitions |
| Sample column | Genotype-level data for one sample |
Inspect the VCF
Keep the original compressed file unchanged.
ls -lh hu24385B.vcf.gz
Count the number of variant rows.
zgrep -vc '^#' hu24385B.vcf.gz
Inspect the metadata.
zgrep '^##' hu24385B.vcf.gz | head -40
Inspect the column header.
zgrep '^#CHROM' hu24385B.vcf.gz
Inspect the first variant rows.
zgrep -v '^#' hu24385B.vcf.gz | head
The first useful skill is recognising the difference between metadata lines, the VCF header, and variant rows.
Check the reference genome
Genomic coordinates depend on the reference genome.
A variant reported at one coordinate in GRCh37 may have a different coordinate in GRCh38. Annotation databases must match the reference build used to generate the VCF.
Check the reference metadata.
zgrep '^##reference=' hu24385B.vcf.gz
Check chromosome naming.
zgrep '^##contig=' hu24385B.vcf.gz | head
A VCF using 1 will not automatically match a BED file using chr1. Coordinate systems and chromosome naming must be consistent before filtering or annotation.
Annotate variants with VEP
A raw VCF usually contains coordinates, alleles, genotype fields, and variant quality information. It does not necessarily contain gene symbols, transcript consequences, protein changes, ClinVar terms, or population frequencies.
Ensembl Variant Effect Predictor, or VEP, adds these fields.
Useful VEP fields include:
| VEP field | Meaning |
|---|---|
| SYMBOL | HGNC gene symbol |
| Gene | Ensembl gene ID |
| Feature | Transcript ID |
| Consequence | Predicted variant consequence |
| HGVSc | Coding DNA change |
| HGVSp | Protein change |
| IMPACT | Broad consequence severity |
| MANE_SELECT | MANE Select transcript flag |
| Existing_variation | Known variant ID, often rsID |
| CLIN_SIG | ClinVar clinical significance where available |
| gnomAD_AF | Population allele frequency where available |
For a small example, the VEP web interface is acceptable. For a whole genome or repeated analysis, run VEP locally with a matching cache.
Split a VCF for web annotation
The VEP web interface has file-size limits. Splitting a VCF is useful for a classroom example. It is not a good production strategy.
The header must be preserved in every split file.
#!/usr/bin/env bash
set -euo pipefail
input="hu24385B.vcf.gz"
outdir="split_vcf"
lines_per_file=150000
mkdir -p "$outdir"
zgrep '^#' "$input" > "$outdir/header.vcf"
zgrep -v '^#' "$input" | split -l "$lines_per_file" - "$outdir/block_"
for block in "$outdir"/block_*; do
cat "$outdir/header.vcf" "$block" > "${block}.vcf"
rm "$block"
done
Each block_*.vcf file now contains the original metadata, the VCF column header, and a subset of variant rows.
Run VEP locally
A local VEP command for a GRCh37 VCF might look like this.
vep \
--input_file hu24385B.vcf.gz \
--output_file hu24385B.vep.tsv \
--cache \
--offline \
--assembly GRCh37 \
--tab \
--symbol \
--canonical \
--mane \
--hgvs \
--protein \
--variant_class \
--sift b \
--polyphen b \
--af_gnomad \
--clin_sig \
--fork 8
For GRCh38, change the assembly argument.
--assembly GRCh38
The output file is a tab-separated table. It can be searched, filtered, joined, and imported into R or Python.
Clean the VEP output
VEP tabular output may include metadata lines beginning with ##. Keep the main header and remove metadata lines.
grep -v '^##' hu24385B.vep.tsv > hu24385B.vep.clean.tsv
Print the column names as a numbered list.
head -1 hu24385B.vep.clean.tsv | tr '\t' '\n' | nl
Find the SYMBOL column.
head -1 hu24385B.vep.clean.tsv | tr '\t' '\n' | nl | grep 'SYMBOL'
Suppose SYMBOL is column 14. Extract unique gene symbols.
awk -F '\t' 'NR > 1 && $14 != "" {print $14}' hu24385B.vep.clean.tsv \
| sort -u > genes_from_vcf.txt
This file contains genes with at least one annotated variant in the VCF.
It is a screening file. It does not contain allele interpretation.
Build a small pharmacogene list
Create a simple pharmacogene file.
CYP2D6
CYP2C19
CYP2C9
VKORC1
TPMT
NUDT15
DPYD
UGT1A1
SLCO1B1
HLA-B
HLA-A
CFTR
G6PD
Save it as:
pharmacogenes.txt
These genes illustrate several pharmacogenomic mechanisms.
| Gene | Common relevance |
|---|---|
| CYP2D6 | Antidepressants, antipsychotics, opioids, beta blockers |
| CYP2C19 | Clopidogrel, proton pump inhibitors, antidepressants |
| CYP2C9 | Warfarin, NSAIDs, phenytoin |
| VKORC1 | Warfarin dose |
| TPMT | Thiopurines |
| NUDT15 | Thiopurines |
| DPYD | Fluoropyrimidines |
| UGT1A1 | Irinotecan and atazanavir |
| SLCO1B1 | Statin-associated myopathy risk |
| HLA-B | Abacavir and allopurinol hypersensitivity |
| HLA-A | Carbamazepine hypersensitivity in some populations |
| CFTR | CFTR modulator eligibility |
| G6PD | Haemolysis risk with oxidant drugs |
The list is deliberately small. A complete clinical pharmacogenomics system would use curated allele definitions and guideline tables.
Compare VCF genes with pharmacogenes
Sort both gene lists.
sort -u genes_from_vcf.txt > genes_from_vcf.sorted.txt
sort -u pharmacogenes.txt > pharmacogenes.sorted.txt
Find genes present in both.
comm -12 genes_from_vcf.sorted.txt pharmacogenes.sorted.txt \
> pharmacogene_hits.txt
Inspect the result.
cat pharmacogene_hits.txt
This step answers one question only:
Which pharmacogenes contain at least one annotated variant in this VCF?
It does not answer:
Does this person have an actionable pharmacogenomic allele?
Filter VEP rows to pharmacogenes
Use the pharmacogene list to retain only VEP rows in relevant genes.
awk 'NR==FNR {genes[$1]; next}
FNR==1 {print; next}
$14 in genes {print}' \
pharmacogenes.txt \
hu24385B.vep.clean.tsv \
> hu24385B.pharmacogene_variants.tsv
This assumes SYMBOL is column 14. Change $14 if your VEP output uses another column position.
Count the remaining rows.
wc -l hu24385B.pharmacogene_variants.tsv
Inspect the first rows.
head hu24385B.pharmacogene_variants.tsv
Reduce the VCF before annotation
A full genome contains millions of variants. If the question is limited to pharmacogenes, reduce the VCF first.
A BED file defines genomic intervals.
chr10 94762681 94855547 CYP2C19
chr22 42126499 42130865 CYP2D6
chr7 117120016 117308718 CFTR
A BED file usually has at least three columns:
| Column | Meaning |
|---|---|
| chrom | Chromosome |
| chromStart | Start coordinate |
| chromEnd | End coordinate |
The BED file must match the VCF reference genome and chromosome naming.
Using bcftools:
bcftools view \
-R pharmacogenes.GRCh37.bed \
-Oz \
-o hu24385B.pharmacogenes.vcf.gz \
hu24385B.vcf.gz
bcftools index hu24385B.pharmacogenes.vcf.gz
Using vcftools:
vcftools \
--gzvcf hu24385B.vcf.gz \
--bed pharmacogenes.GRCh37.bed \
--recode \
--keep-INFO-all \
--out hu24385B.pharmacogenes
The reduced VCF is faster to annotate and easier to inspect.
Prepare a drug-gene table
A simple drug-gene table can be stored as tab-separated text.
Gene.Name Drug.ID Drug.Name Relationship Source Evidence
CYP2C19 DB00758 Clopidogrel metabolism CPIC guideline
CYP2D6 DB00472 Fluoxetine metabolism PharmGKB curated
SLCO1B1 DB00641 Simvastatin toxicity CPIC guideline
DPYD DB00544 Fluorouracil toxicity CPIC guideline
CFTR DB08820 Ivacaftor target FDA label
This file is a teaching substitute for a curated pharmacogenomic database.
In a professional setting, the equivalent table would be assembled from controlled sources such as CPIC, PharmGKB, PharmVar, DPWG, FDA labels, and internal validation records.
Merge VEP and drug-gene data in R
Import the VEP output and drug-gene table.
vep <- read.delim(
"hu24385B.vep.clean.tsv",
stringsAsFactors = FALSE,
check.names = FALSE
)
drug_genes <- read.delim(
"drug_gene_table.tsv",
stringsAsFactors = FALSE,
check.names = FALSE
)
Rename the VEP gene symbol column to match the drug-gene table.
names(vep)[names(vep) == "SYMBOL"] <- "Gene.Name"
Remove rows without a gene symbol.
vep <- vep[!is.na(vep$Gene.Name) & vep$Gene.Name != "", ]
Merge by gene name.
merged <- merge(
x = vep,
y = drug_genes,
by = "Gene.Name",
all = FALSE
)
Remove low-information variant consequences for this exercise.
low_information <- paste(
c(
"synonymous_variant",
"intron_variant",
"upstream_gene_variant",
"downstream_gene_variant",
"UTR",
"non_coding_transcript",
"NMD_transcript_variant"
),
collapse = "|"
)
keep <- !grepl(low_information, merged$Consequence)
merged_filtered <- merged[keep, ]
Write the candidate table.
write.table(
merged_filtered,
file = "pharmacogene_variant_drug_matches.tsv",
sep = "\t",
quote = FALSE,
row.names = FALSE
)
The output table links annotated variants to drug-gene records.
It is still a candidate table. Interpretation comes later.
Merge VEP and drug-gene data in Python
The same join can be written in Python using pandas.
import pandas as pd
vep = pd.read_csv("hu24385B.vep.clean.tsv", sep="\t")
drug_genes = pd.read_csv("drug_gene_table.tsv", sep="\t")
vep = vep.rename(columns={"SYMBOL": "Gene.Name"})
vep = vep[vep["Gene.Name"].notna() & (vep["Gene.Name"] != "")]
merged = vep.merge(drug_genes, on="Gene.Name", how="inner")
low_information = (
"synonymous_variant|"
"intron_variant|"
"upstream_gene_variant|"
"downstream_gene_variant|"
"UTR|"
"non_coding_transcript|"
"NMD_transcript_variant"
)
merged_filtered = merged[
~merged["Consequence"].astype(str).str.contains(low_information, regex=True)
]
merged_filtered.to_csv(
"pharmacogene_variant_drug_matches.tsv",
sep="\t",
index=False
)
The Python version is shorter. The Bash and R versions make more of the file mechanics visible.
Read the candidate table
The candidate table should contain enough fields to show the chain from variant to drug relationship.
| Column | Meaning |
|---|---|
| Gene.Name | HGNC gene symbol |
| Location or Uploaded_variation | Variant coordinate or VEP variant ID |
| Allele | Alternate allele |
| Consequence | VEP consequence |
| HGVSc | Coding change |
| HGVSp | Protein change |
| Existing_variation | Known ID such as rsID |
| CLIN_SIG | ClinVar assertion where available |
| Drug.ID | Drug identifier |
| Drug.Name | Drug name |
| Relationship | Metabolism, toxicity, efficacy, target, biomarker, or warning |
| Source | CPIC, PharmGKB, FDA, DrugBank, or other |
| Evidence | Guideline, label, curated evidence, prediction, or candidate match |
A candidate row does not mean that a drug should be given or avoided. It means the row has enough structure for review.
Consequence is not interpretation
VEP consequence terms describe predicted molecular effect.
| Consequence | Typical meaning |
|---|---|
| stop_gained | Creates a premature stop codon |
| frameshift_variant | Changes the reading frame |
| splice_acceptor_variant | Affects a canonical splice acceptor |
| splice_donor_variant | Affects a canonical splice donor |
| start_lost | Alters the start codon |
| missense_variant | Changes one amino acid |
| synonymous_variant | Does not change the amino acid sequence |
| intron_variant | Located in an intron |
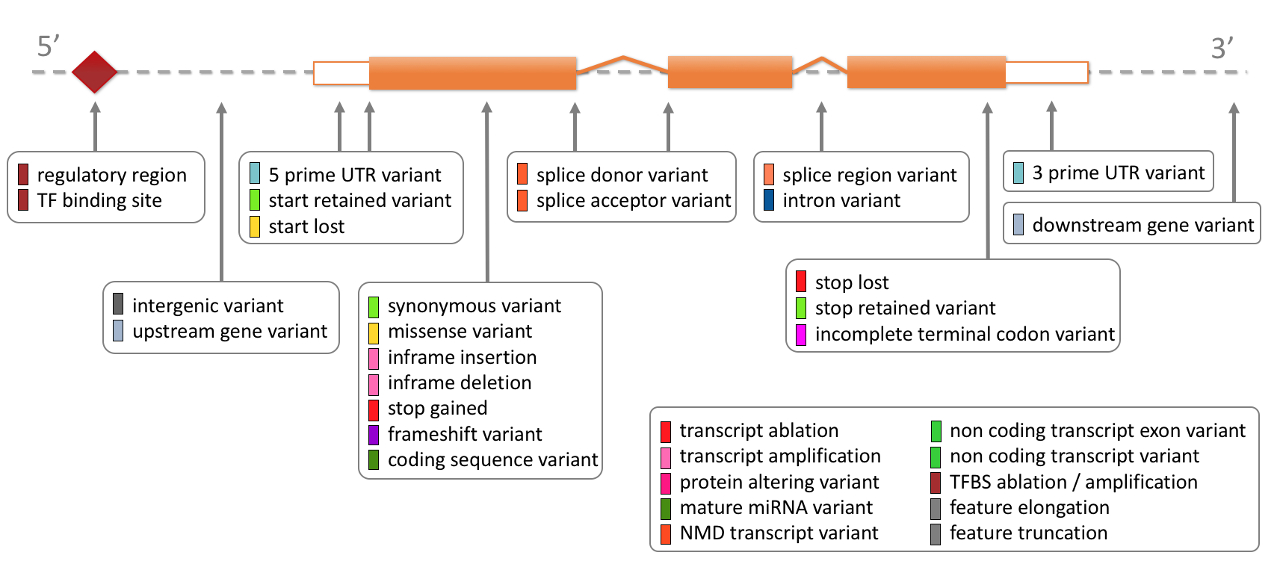
A high-impact consequence can support interpretation. It is not automatically pathogenic. A missense variant can be benign, damaging, uncertain, or irrelevant to the drug response.
Transcript choice also matters. A variant can be coding on one transcript and non-coding on another. MANE Select transcripts are often preferred for clinical reporting where available.
Known pharmacogenomic alleles
Many pharmacogenomic results are not interpreted from single VCF rows.
They are often interpreted as star alleles, diplotypes, copy number states, repeat genotypes, or HLA alleles.
| Gene | Result type |
|---|---|
| CYP2D6 | Star alleles, copy number, hybrid alleles |
| CYP2C19 | Star alleles |
| CYP2C9 | Star alleles |
| TPMT | Star alleles or named variants |
| NUDT15 | Named alleles |
| HLA-B | HLA allele, such as HLA-B*57:01 |
| UGT1A1 | Repeat genotype, such as UGT1A1*28 |
| SLCO1B1 | Specific variant or haplotype |
A standard SNV VCF can miss important pharmacogenomic structure.
| Task | Tools or resources |
|---|---|
| CYP2D6 calling | Aldy, Stargazer, Cyrius, Astrolabe |
| HLA typing | OptiType, HLA-HD, HISAT-genotype |
| Pharmacogenomic annotation | PharmCAT |
| Star allele definitions | PharmVar |
| Clinical recommendations | CPIC, DPWG |
Gene-level matching is the first pass. Allele-level interpretation is required before a pharmacogenomic conclusion.
Zygosity and dosage
Genotype affects interpretation.
| State | Meaning |
|---|---|
| Heterozygous | One alternate allele |
| Homozygous | Two alternate alleles |
| Hemizygous | One copy of the region |
| Compound heterozygous | Two different variants in the same gene |
| Copy number change | Deletion or duplication of a gene or region |
| Mosaic | Variant present in only some cells |
Pharmacogenomic reports often translate genotype into a functional phenotype, such as poor, intermediate, normal, rapid, or ultrarapid metaboliser.
CYP2D6 shows why dosage matters. A person may carry inactive alleles, reduced-function alleles, gene duplications, or hybrid alleles. A simple SNV table cannot always resolve that structure.
Drug-gene relationship types
A drug-gene match can mean several things.
| Relationship | Meaning |
|---|---|
| Metabolism | The gene product affects drug exposure |
| Toxicity | The genotype changes adverse event risk |
| Efficacy | The genotype changes probability of response |
| Target | The drug binds the gene product |
| Biomarker | The gene or variant is part of a labelled indication |
| Warning | The genotype or disease state changes risk |
A drug target is not the same as a prescribing rule.
A drug may bind a protein encoded by a gene, but inherited variation in that gene may have no known treatment effect. A pharmacogenomic variant may affect metabolism of many drugs without being part of the drug target.
The FDA pharmacogenomic biomarker table and drug prescribing information help distinguish these categories.
Unknown variants
Most variants in a genome are not actionable.
A rare missense variant in a pharmacogene may be known, benign, uncertain, technical, or research-only.
| Interpretation | Meaning |
|---|---|
| Known actionable allele | Evidence supports a drug-specific recommendation |
| Known benign or tolerated variant | No action expected |
| Variant of uncertain significance | Insufficient evidence |
| Technical artefact | Variant call is not reliable |
| Research candidate | Interesting, but not clinically interpretable |
Prediction tools can help rank variants. They do not define clinical action.
| Tool | Use |
|---|---|
| CADD | General deleteriousness score |
| REVEL | Missense variant prediction |
| AlphaMissense | Missense effect prediction |
| SIFT | Protein tolerance prediction |
| PolyPhen-2 | Missense effect prediction |
| SpliceAI | Splice effect prediction |
Predicted effects should remain labelled as predictions.
Limits of the first-principles workflow
The workflow explains the mechanics of a simple pharmacogenomics analysis. It does not replace a validated clinical pipeline.
Special caution is needed for:
| Gene or region | Reason |
|---|---|
| CYP2D6 | Copy number, hybrid alleles, and complex haplotypes |
| HLA genes | High polymorphism and specialised typing requirements |
| UGT1A1 | Repeat alleles may not be represented in simple VCFs |
| Structural variants | Often absent from SNV-focused VCFs |
| Low coverage regions | Negative results may be uninformative |
| Direct-to-consumer data | Often incomplete for clinical PGx |
| Transcript ambiguity | Consequence can change by transcript |
| Reference mismatch | Coordinates and annotations can become incorrect |
The most common error is treating a matched gene as an interpreted result.
The correct interpretation chain is stricter.
variant call
↓
quality check
↓
correct reference build
↓
correct transcript or allele definition
↓
validated pharmacogenomic allele
↓
drug-specific guideline or label
↓
clinical interpretation
Practical summary
This exercise builds a candidate pharmacogenomics table from first principles.
It uses:
VCF
VEP annotation
gene extraction
pharmacogene matching
drug-gene joining
consequence filtering
candidate review
The file operations are straightforward. The interpretation is not.
A candidate table can show where to look. A pharmacogenomic conclusion requires allele-level evidence, drug-specific guidance, and quality-controlled genotyping.
References and resources
- CPIC: Clinical Pharmacogenetics Implementation Consortium, https://cpicpgx.org
- PharmGKB: Pharmacogenomics Knowledgebase, https://www.pharmgkb.org
- PharmVar: Pharmacogene Variation Consortium, https://www.pharmvar.org
- DPWG: Dutch Pharmacogenetics Working Group, https://www.pharmgkb.org/page/dpwg
- FDA pharmacogenomic biomarkers in drug labelling, https://www.fda.gov/drugs/science-and-research-drugs/table-pharmacogenomic-biomarkers-drug-labeling
- Ensembl Variant Effect Predictor, https://www.ensembl.org/info/docs/tools/vep/index.html
- MANE Select, https://www.ncbi.nlm.nih.gov/refseq/MANE/
- DrugBank, https://go.drugbank.com
- gnomAD, https://gnomad.broadinstitute.org
- ClinVar, https://www.ncbi.nlm.nih.gov/clinvar
- Relling MV, Evans WE. Pharmacogenomics in the clinic. Nature. 2015;526:343-350. doi: 10.1038/nature15817
- Yip VL, Hawcutt DB, Pirmohamed M. Pharmacogenetic markers of drug efficacy and toxicity. Clin Pharmacol Ther. 2015;98:61-70. doi: 10.1002/cpt.135
- Adams DR, Eng CM. Next-generation sequencing to diagnose suspected genetic disorders. N Engl J Med. 2018. doi: 10.1056/NEJMra1711801
Maintained by Dylan Lawless. Scientist at EPFL. Resume and Google scholar. RSS feed