Variant annotation
Variant annotation is a critical step in clinical genetics. Popular tools for applying annotation data to VCF format genetic data include:
- NIRVANA https://illumina.github.io/NirvanaDocumentation/
- ANNOVAR https://annovar.openbioinformatics.org/en/latest/
- Variant Effect Predictor (VEP) http://grch37.ensembl.org/Homo_sapiens/Tools/VEP/
Additionally, these tools require a set of data sources containing the annotation information which will be applied to each variant. However, each tool generally provides a ready-to-go database that can be utilised.
As an example of function, Variant Effect Predictor (VEP) is very useful. A real-world example of VEP in use can be read in this pharmacogenomics article (written for my students). During variant annotation, VEP supplies a “consequence” column. Consequences are general and based on translation of genetic code in humans. The loss-of-function (LoF) consequence is the simplest example to interpret (splice mutation, stop mutation, etc.). The variant consequence may be one of the defining criteria by which variants can be included in analysis since they are interpretable or of ostensibly known significance.
NB: Correctly identifying a single disease-causing variant for monogenic disease is much more complex than simply selecting the highest impact consequence. However, this example is useful as one step in the multi-step process.
Note: Using consequence alone could introduce spurious results so it is best to have solid criteria for selecting consequences of interest combined with additional filtering methods. See the ACMG interpretation standards for examples.
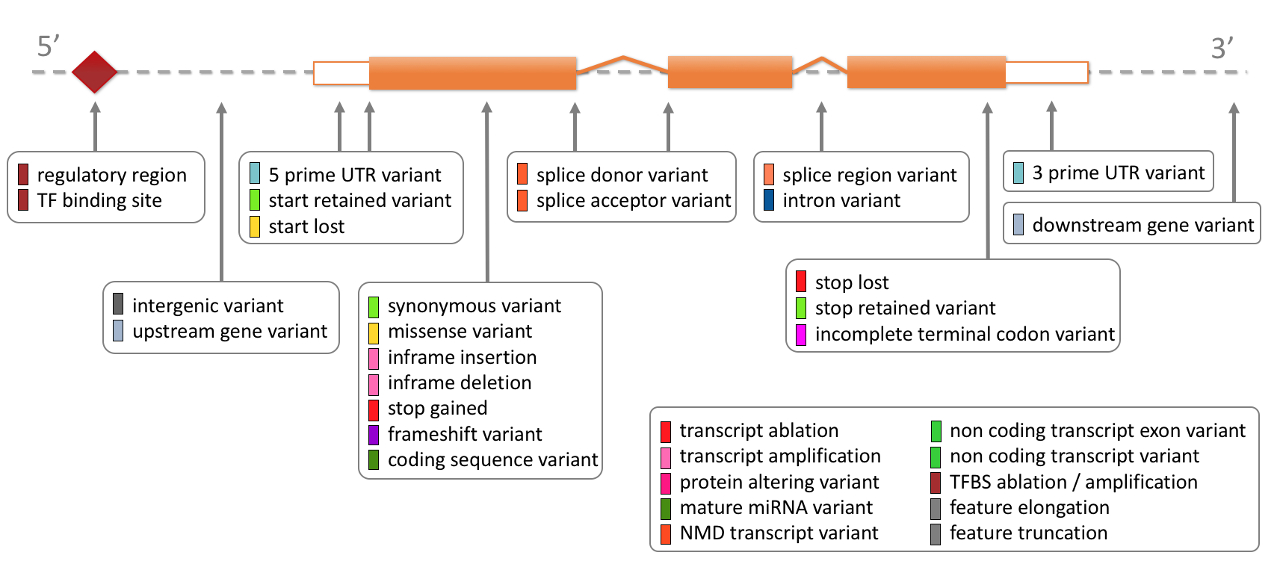
The consequences provided by VEP are too long to discuss in detail here.
The table from the ensembl website is worth reading.
HIGH impact variants
might be a likely consequence for identifying candidates disease-causing:
Ensembl Variation - Calculated variant consequences.
Note: For working product, the code should be run offline (a perl program with a few local library dependencies). The databases/cache that it uses are a bit too large to include on in a user software. In the real world you would have to send anonymised packets from the user via an API for accessing the genomic databases hosted on your servers. Make sure to check their license to see if you can use oftware and databases in a commercial product. http://www.ensembl.org/info/about/legal/code_licence.html
Running the software:
- Using VEP is a vital part of converting the DNA variant information (genome position and nucleotide change) into annotated variant effects (protein coding change, gene name, predicted pathogenicity).
- It requires the VEP code to run and requires a copy of the database files (reference genome, gene information, etc.).
- You can upload a small number of variants to the online VEP web server to do this, or you can download the database and code to run on your own computer/server.
The terms in the table below are shown in order of severity (more severe to less severe) as estimated by Ensembl. In single case studies, we often find rare disease due to variants of “IMPACT” that is “HIGH or MODERATE”. However, there may be a bias in that these variants are often most likely to be functionally interpretable. Variants with effect that is more difficult to predict might only be detected in large-scale association studies where a is statistically valid explanation for correlation/cause is possible. Other tools may provide alternative estimates than ones shown here from VEP. Missense variants may have further annotation based on their effect for protein function, using a number of algorithms (see VEP Ensembl Variation - Calculated variant consequences).
Consequence table
Maintained by Dylan Lawless. Scientist at EPFL. Resume and Google scholar. RSS feed